- Shwachman-Bodian-Diamond-Syndrom
-
Klassifikation nach ICD-10 Q45.0 Sonstige angeborene Fehlbildungen des Verdauungssystems ICD-10 online (WHO-Version 2011) Das Shwachman-Bodian-Diamond-Syndrom (SBDS) ist eine seltene angeborene Erkrankung, die durch eine mangelnde Bildung von Verdauungsenzymen in der Bauchspeicheldrüse (Exokrine Pankreasinsuffizienz), Störungen der Funktion des Knochenmarkes mit einer Neigung zur Entwicklung einer Leukämie, Skelettfehlbildungen und Minderwuchs gekennzeichnet ist. Nach der Mukoviszidose ist es die zweithäufigste Ursache für eine exokrine Pankreasinsuffizienz im Kindesalter.
Inhaltsverzeichnis
Geschichte
Die Krankheit wurde erstmals durch Bodian und Kollegen[1] im Jahre 1964 erkannt und im gleichen Jahr durch Shwachman und Diamond beschrieben.[2] 2001 wurde durch eine Linkage-Analyse in mehreren SBDS-Familien gezeigt, dass der gesuchte Genlocus auf Chromosom 7 liegen muss. Ein Jahr später wurde die genaue Region in der Nähe des Centromers definiert und später die Assoziation des SBDS-Gen[3] mit der Krankheit nachgewiesen.
Genetik
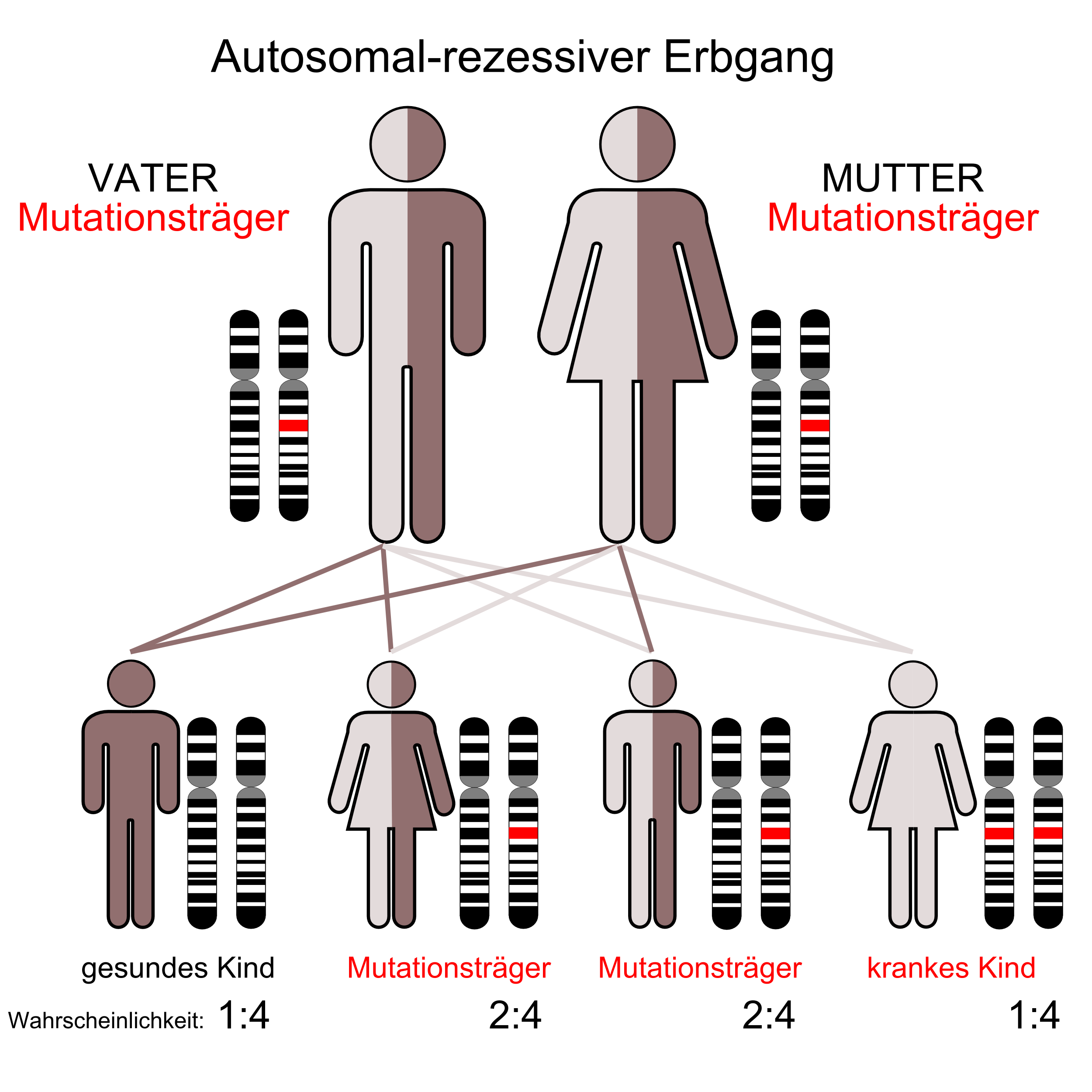
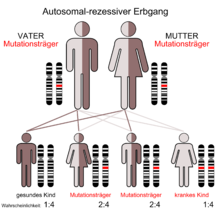 Das Shwachman-Diamond-Syndrom hat einen autosomal-rezessiven Erbgang.
Das Shwachman-Diamond-Syndrom hat einen autosomal-rezessiven Erbgang.
Das SBDS ist charakterisiert durch einen autosomal-rezessiven Vererbungsmodus. Das verursachende Gen (SBDS-Gen) liegt auf dem Chromosom 7 an Position 7q11. Es besteht aus 5 Exons, die für eine 1,6 kB große mRNA codieren. Das SBDS-Gen liegt in einer duplizierten Region. Die zu 97 % identische Kopie ist nicht funktional, da sie eine Reihe von inaktivierenden Mutationen enthält und wird daher als Pseudogen angesehen. Bei einer Untersuchung von 158 Familien fand man, dass etwa 3/4 der krankheitsbezogenen Mutationen Folge einer Genkonversion sind und etwa 90 Prozent der Betroffenen solche Mutationen aufwiesen. Eine Genkonversion findet statt, wenn intaktes Gen und Pseudogen bei der Meiose rekombinieren und dabei mutierte Sequenzen des Pseudogens in das funktionale Gen einkopiert werden und dieses so inaktivieren. Die häufigsten beim SBDS-Gen vorkommenden Konversionen betreffen eine Mutation einer Splice-Site und eine Nonsense-Mutation im Exon 2.
Pathophysiologie
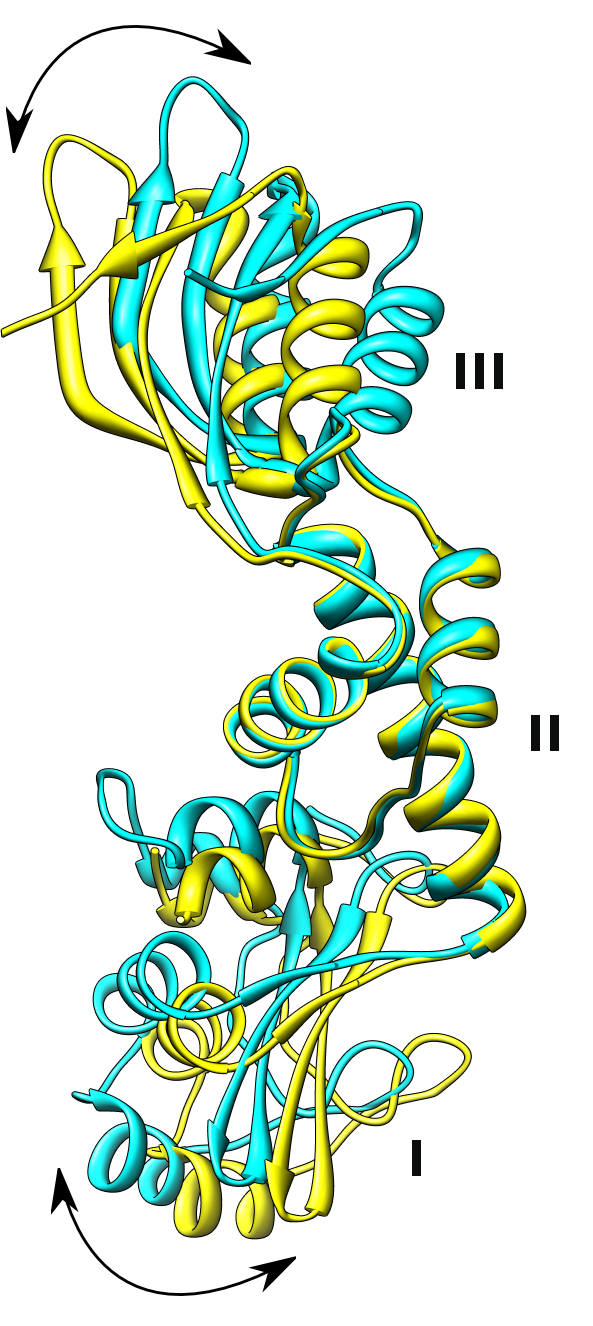
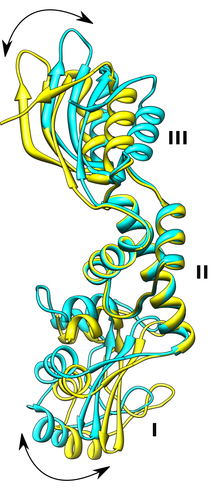 Bändermodell des SBDS-Proteins mit den Domänen I, II und III.[4]
Bändermodell des SBDS-Proteins mit den Domänen I, II und III.[4]Das SBDS-Gen wird in allen Geweben exprimiert und codiert für ein Protein mit 250 Aminosäuren. Die Funktion ist nicht bekannt. Bislang kennt man keine Sequenzhomologie zu irgendeinem anderen Gen, aus der sich die Funktion erschließen ließe. Die dreidimensionale Struktur eines zum SBDS-Gen orthologen Protein von Archaeen wurde aufgeklärt und man fand, dass die meisten Mutationen beim Menschen eine Region betreffen, die eine ungewöhnliche Struktur am N-terminalen Ende des Proteins betreffen. Es gibt eine Reihe von Hinweisen dafür, dass das SBDS-Protein beim Zusammenbau von Ribosomen eine Rolle spielt. Die weite Verbreitung des Proteins in allen Geweben und bei Eukaryoten und Archäen spricht dafür, dass es an einem sehr fundamentalen und evolutionär hochkonservierten biologischen Prozess beteiligt ist. Für eine Beteiligung des SBDS-Proteins am Stoffwechsel oder Zusammenbau der Ribosomen spricht auch die Lokalisation des Proteins im Nukleolus. Bislang ist nicht klar, wieso die Störung eines sehr fundamentalen zellulären Prozesses sehr spezifische Störungen verursacht. Allerdings findet man eine ähnliche Konstellation auch beim Diamond-Blackfan-Syndrom, der x-chromosomalen Dyskeratosis congenita und der Knorpel-Haar-Hypoplasie. Drei Erkrankungen, bei denen auch ein Zusammenhang mit defekten Ribosomen-Funktionen vermutet wird.
Klinisches Bild
Die von dieser Erkrankung betroffenen Patienten zeigen eine Vielzahl von Störungen und Symptomen. Man findet üblicherweise die Trias exokrine Pankreasinsuffizienz, Leukopenie und Skelettveränderungen.[5] Eine Neutropenie (Sonderform der Leukopenie) kann zeitweise (intermittierend) oder dauerhaft vorkommen. Der niedrige Neutrophilenanteil kann mit einem erhöhten Infektionsrisiko einhergehen und für die Patienten lebensbedrohlich sein. Auch eine Anämie (Blutarmut) und ein Blutplättchenmangel (Thrombozytopenie) können auftreten. Die Ursache für diese Blutbildveränderungen ist ein Abnahme der Zellen im Knochenmark, das einen Abbruch der Zellreifung bei den myeloischen Zellen zeigt. Die Patienten können in der Folge einen zunehmenden Ausfall der Konchenmarksfunktion erleiden oder eine Akute myeloische Leukämie entwickeln.
Die exokrine Pankreasinsuffizienz ist eine Folge des Verlustes der Azinuszellen der Bauchspeicheldrüse, die die Verdauungsenzyme herstellen. Diese nehmen in ihrer Anzahl ab und werden durch Fettgewebe ersetzt. Der Mangel an Verdauungsenzymen führt zu einer Störung der Fettverdauung mit der Folge eines Malabsorptionssyndroms. Im Gegensatz zu anderen Störungen mit einer angeborenen exokrinen Pankreasinsuffizienz – beispielsweise das Johanson-Blizzard-Syndrom, bei dem die Funktionsstörung der Bauspeicheldrüse einen progressiven Charakter hat – kann sich beim SDS die Bauchspeicheldrüsenfunktion mit der Zeit verbessern.
Mehr als die Hälfte der Patienten zeigen einen Minderwuchs, der nicht im Zusammenhang mit dem Ernährungsstatus steht. Die Skelettveränderungen betreffen bei jedem zweiten Patienten eine metaphysäre Dysostose, ebenso häufig Veränderungen des Brustkorbes, zum Beispiel mit verkürzten Rippen. Die Skelettveränderungen sind ein sehr variables Symptom. Sie scheinen aber bei allen Patienten mit einer gesicherten Diagnose vorzukommen, wenn auch in sehr unterschiedlicher Ausprägung. Dabei sind die Symptome bei Individuen mit identischem Genotyp unterschiedlich. Eine Genotyp-Phänotyp-Beziehung konnte nicht beobachtet werden.[6]
Diagnose
Bei Feststellung der ersten Symptome kann das SBDS Ähnlichkeiten mit der Mukoviszidose aufweisen. Eine Mukoviszidose kann aber durch einen normalen Schweißtest ausgeschlossen werden. Die unterschiedliche Ausprägung der Symptome, deren vorübergehender Charakter und die Tatsache, dass sich manche Beschwerden mit der Zeit bessern, machen es manchmal schwierig ein SBDS zu diagnostizieren. Dieses kann sich entweder mit einem Malabsorptionssyndrom oder mit Blutbildveränderungen präsentieren. Bei manchen Patienten stehen die Skelettveränderungen im Vordergrund, einschließlich Deformationen der Rippen, die zu Atembeschwerden führen können. Üblicherweise wird die Verdachtsdiagnose bei Vorliegen einer exokrinen Pankreasinsuffizinez mit Blutbildstörungen im Kindesalter gestellt. Die Skelettveränderung und Minderwuchs können die Diagnose stützen. Da das für die Erkrankung verantwortliche Gen identifiziert ist, kann eine genetische Diagnostik erfolgen, obwohl dadurch eine sorgfältige klinische Diagnose nicht überflüssig wird.
Behandlung
Die exokrine Pankreasinsuffizienz kann man durch eine Substitution der Pankreasenzyme behandeln und die Skelettabnormitäten können chirurgische Eingriffe notwendig machen. Eine Neutropenie kann mit G-CSF behandelt werden. Befürchtungen, dass die Behandlung mit G-CSF das Leukämierisiko erhöht, konnten nicht bestätigt werden.[7] Falls die Patienten eine progressive Knochenmarkdepression zeigen, kann eine Stammzelltransplantation notwendig sein. Allerdings sollen Patienten mit einem SBDS ein erhöhtes Risiko zur Entwicklung einer Graft-versus-Host-Reaktion haben.[8] Von der Untersuchung der dem SBDS zugrundeliegenden genetischen Störung erhofft man sich ein verbessertes Verständnis der molekularen Ursachen der Erkrankung und damit möglicherweise neue therapeutische Möglichkeiten.
Verwendete Literatur
- Shammas C, Menne TF, Hilcenko C, Michell SR, Goyenechea B, Boocock GR, Durie PR, Rommens JM, Warren AJ: Structural and mutational analysis of the SBDS protein family. Insight into the leukemia-associated Shwachman-Diamond Syndrome.. In: J Biol Chem.. 280, Nr. 19, 2005, S. 19221–9. doi:10.1074/jbc.M414656200. PMID 15701631.
- Cesaro S, Oneto R, Messina C, Gibson BE, Buzyn A, Steward C, Gluckman E, Breddius R, Boogaerts M, Vermylen C, Veys P, Marsh J, Badell I, Michel G, Gungor T, Niethammer D, Bordigoni P, Oswald C, Favre C, Passweg J, Dini G: Haematopoietic stem cell transplantation for Shwachman-Diamond disease: a study from the European Group for blood and marrow transplantation.. In: Br J Haematol. 31, Nr. 2, 2005, S. 231–6. doi:10.1111/j.1365-2141.2005.05758.x. PMID 16197455.
- Donadieu J, Michel G, Merlin E, Bordigoni P, Monteux B, Beaupain B, Leverger G, Laporte JP, Hermine O, Buzyn A, Bertrand Y, Casanova JL, Leblanc T, Gluckman E, Fischer A, Stephan JL: Hematopoietic stem cell transplantation for Shwachman-Diamond syndrome: experience of the French neutropenia registry.. In: Bone Marrow Transplant. 36, Nr. 9, 2005, S. 787–92. doi:10.1038/sj.bmt.1705141. PMID 16151425.
- Austin KM, Leary RJ, Shimamura A: The Shwachman-Diamond SBDS protein localizes to the nucleolus.. In: Blood. 106, Nr. 4, 2005, S. 1253–8. doi:10.1182/blood-2005-02-0807. PMID 15860664.
- Makitie O, Ellis L, Durie PR, Morrison JA, Sochett EB, Rommens JM, Cole WG: Skeletal phenotype in patients with Shwachman-Diamond syndrome and mutations in SBDS.. In: Clin Genet. 65, Nr. 2, 2004, S. 101–12. doi:10.1111/j.0009-9163.2004.00198.x. PMID 14984468.
- Boocock GRB, Morrison JA, Popovic M, Richards N, Ellis L, Durie PR, Rommens JM: Mutations in SBDS are associated with Shwachman-Diamond syndrome. In: Nat Genet. 33, Nr. 1, 2003, S. 97–101. doi:10.1038/ng1062. PMID 12496757.
- Popovic M, Goobie S, Morrison J, Ellis L, Ehtesham N, Richards N, Boocock G, Durie PR, Rommens JM: Fine mapping of the locus for Shwachman-Diamond syndrome at 7q11, identification of shared disease haplotypes, and exclusion of TPST1 as a candidate gene.. In: Eur J Hum Genet. 10, Nr. 4, 2002, S. 250–8. doi:10.1038/sj.ejhg.5200798. PMID 12032733.
- Smith OP: Shwachman-Diamond syndrome.. In: Semin Hematol. 39, Nr. 2, 2002, S. 95–102. doi:10.1053/shem.2002.31915. PMID 11957191.
- Goobie S, Popovic M, Morrison J, Ellis L, Ginzberg H, Boocock GR, Ehtesham N, Betard C, Brewer CG, Roslin NM, Hudson TJ, Morgon K, Fujiwara TM, Durie PR, Rommens JM: Shwachman-Diamond syndrome with exocrine pancreatic dysfunction and bone marrow failure maps to the centromeric region of chromosome 7.. In: Am J Hum Genet. 68, Nr. 4, 2001, S. 1048–54. doi:10.1086/319505. PMID 11254457.
- Cipolli M: Shwachman-Diamond syndrome: clinical phenotypes.. In: Pancreatology. 1, Nr. 5, 2001, S. 543–8. doi:10.1159/000055858. PMID 12120235.
- Cipolli M, D’Orazio C, Delmarco A, Marchesini C, Miano A, Mastella G: Shwachman’s syndrome: pathomorphosis and long-term outcome.. In: J Pediatr Gastroenterol Nutr. 29, Nr. 3, 1999, S. 265–72. doi:10.1097/00005176-199909000-00006. PMID 10467990.
- Ginzberg H, Shin J, Ellis L, Morrison J, Ip W, Dror Y, Freedman M, Heitlinger LA, Belt MA, Corey M, Rommens JM, Durie PR: Shwachman syndrome: phenotypic manifestations of sibling sets and isolated cases in a large patient cohort are similar.. In: J Pediatr. 135, Nr. 1, 1999, S. 81–8. doi:10.1016/S0022-3476(99)70332-X. PMID 10393609.
- Michels VV, Donovan GK: Shwachman syndrome: unusual presentation as asphyxiating thoracic dystrophy.. In: Birth Defects Orig Artic Ser. 18, Nr. 3B, 1982, S. 129–34. PMID 7139093.
- Shwachman H, Diamond LK, Oski FA, Khaw KT: The syndrome of pancreatic insufficiency and bone marrow dysfunction.. In: J Pediatr. 65, 1964, S. 645–63. doi:10.1016/S0022-3476(64)80150-5. PMID 14221166.
- Bodian M, Sheldon W, Lightwood R: Congenital hypoplasia of the exocrine pancreas.. In: Acta Paediatr. 53, 1964, S. 282–93. doi:10.1111/j.1651-2227.1964.tb07237.x. PMID 14158482.
- Einzelnachweise
- ↑ M. Bodian u. a.: Congenital hypoplasia of the exocrine pancreas. In: Acta Paediat 53, 1964, S. 282–293. PMID 14158482
- ↑ H. Shwachman, L. Diamond u. a.: The syndrome of pancreatic insufficiency and bone marrow dysfunction. In: J Pediat 65, 1964, S. 645–663. PMID 14221166
- ↑ ensembl.org: Gene: SBDS (ENSG00000126524) Shwachman-Bodian-Diamond syndrome. Abgerufen am 26. Juni 2010
- ↑ C Leong Ng, David G Waterman, Eugene V Koonin, Alison D Walters, James PJ Chong, Michail N Isupov, Andrey A Lebedev, David HJ Bunka, Peter G Stockley, Miguel Ortiz-Lombardía and Alfred A Antson: Conformational flexibility and molecular interactions of an archaeal homologue of the Shwachman-Bodian-Diamond syndrome protein. In: BMC Structural Biology 2009, 9:32 doi:10.1186/1472-6807-9-32 (Open Access)
- ↑ Hall GW, Dale P, Dodge JA. Shwachman-Diamond syndrome: UK perspective. in: Arch Dis Child. 2006 Jun;91(6):521-4. PMID 16714727
- ↑ Mäkitie O, Ellis L, Durie PR, Morrison JA, Sochett EB, Rommens JM, Cole WG. Skeletal phenotype in patients with Shwachman-Diamond syndrome and mutations in SBDS. in: Clin Genet. 2004 Feb;65(2):101-12. PMID 14984468
- ↑ Confer/Miller (2007): Long-term safety of filgrastim (rhG-CSF) administration In: British Journal of Haematology 137(1): 76–80, PMCID: PMC1920544 [1]
- ↑ Burroughs L, Woolfrey A, Shimamura A. Shwachman-Diamond syndrome: a review of the clinical presentation, molecular pathogenesis, diagnosis, and treatment. Hematol Oncol Clin North Am. 2009 Apr;23(2):233-48. PMID 19327581.
Weblinks
- Shwachman-Bodian-Diamond-Syndrom bei Online Mendelian Inheritance in Man
- Shwachman-Bodian-Diamond-Syndrom bei Orphanet (Datenbank für seltene Krankheiten)
- GeneReviews/NCBI/NIH/UW entry on Shwachman-Diamond Syndrome
- Shwachman-Diamond Syndrome research study of Inherited Bone Marrow Failure Syndromes (IBMFS)
- Shwachman-Diamond Syndrome Foundation

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Krankheitsbild in der Kinderheilkunde
- Erbkrankheit
- Krankheitsbild in der Gastroenterologie
- Krankheitsbild in Hämatologie und Onkologie
Wikimedia Foundation.