- I-Zellkrankheit
-
Die I-Zellkrankheit, auch als Mukolipidose II bezeichnet, ist eine sehr seltene autosomal rezessiv vererbte lysosomale Speicherkrankheit.
Klassifikation nach ICD-10 E77.0 Defekte der posttranslationalen Modifikation lysosomaler Enzyme
- Mukolipidose II [I-Zell-Krankheit]ICD-10 online (WHO-Version 2011) Inhaltsverzeichnis
Entdeckung
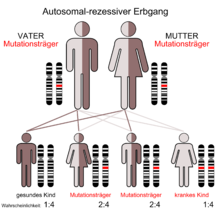 Autosomal-rezessiver Erbgang
Autosomal-rezessiver Erbgang
Leroy und DeMars beschrieben 1967 erstmals die I-Zellkrankheit, die der Mukopolysaccharidose Typ I (Morbus Hurler) ähnelt. Die Patienten zeigten jedoch keine Mukopolysaccharid-Ausscheidung. Dafür konnten die beiden Forscher in den Fibroblasten der Haut Einschlusskörperchen (engl.: inclusin-cells) nachweisen. Die Einschlüsse gaben der Krankheit ihren englischen Namen I-Cell-Disease.[1]
Die Krankheit tritt ausgesprochen selten auf. Die Inzidenz der beiden Mukolipidosen II und III beträgt zusammen ca. 0,3 : 100.000.[2]
Ursache
Die Ursache der Krankheit ist eine mangelnde Aktivität des Enzyms N-Acetylglucosaminyl-1-Phosphotransferase (EC 2.7.8.17), die bewirkt, dass ein großer Teil lysosomaler Enzyme nicht in das Innere der Lysosomen gelangen kann. Die Zielsteuerung lysosomaler Enzyme ist durch einen Defekt der Phosphotransferase, die normalerweise die Synthese eines spezifischen Sortierungssignals ermöglicht, gestört. Die Markierung mit Mannose-6-phosphat kann nicht stattfinden und die lysosomalen Enzyme können nicht mehr sortiert werden. Sie gelangen unkontrolliert über die Plasmamembran in die extrazelluläre Matrix.[3]
Symptome
Die Symptome der Krankheit sind meist schon bei der Geburt, spätestens wenige Monate nach der Geburt, sichtbar. Die Symptome gleichen denen des Hurler-Syndroms (Mukopolysaccharidose I), sind aber in der Regel früher zu beobachten. Zudem zeigen die Patienten keine Mukopolysaccharid-Ausscheidungen. Die Symptome zeigen von Familie zu Familie und auch innerhalb einer Familie, eine große Vielfalt an Varianten.[4]
Nach Kornfeld und Sly sind folgende klinischen Merkmale möglich:[5]
1. Skelett
Kyphoskoliose, Hüftluxation, Klumpfüße, Gelenkkontrakturen, Wirbeldeformitäten, Minderwuchs, Dysostosis multiplex2. Innere Organe
Hepatosplenomegalie, Kardiomegalie, Herzvitien3. Gesicht und Kopf
Vergröberte Gesichtszüge, Exophthalmus, hyperplastisches Zahnfleisch, Skaphozephalie, offen gehaltener Mund, tief eingesunkene Nasenwurzel4. Augen
Hornhauttrübungen, geschwollene Augenlider5. Haut
Dicke derbe Haut6. ZNS
Schwere psychomotorische und geistige RetardierungDiagnose
Durch eine biochemische Bestimmung der Aktivität der lysosomalen Enzyme im Serum, kann die Diagnose gestellt werden. Das Verhältnis von intra- und extrazellulärer Aktivität der lysosomalen Enzyme und die Aktivität der Phosphotransferase in kultivierten Fibroblasten, sind weitere mögliche Laborparameter.[4]
Die Einschlüsse in den Fibroblasten sind Anreicherungen von Mukopolysacchariden, Lipiden und Oligosacchariden.[6][7][8]
Genetik
Das Enzym N-Acetyl-Glucosamin-1-Phosphotransferase besteht aus drei Untereinheiten (alpha, beta und gamma), die von zwei verschiedenen Genen kodiert werden. Ein Gen kodiert dabei die α- und β-, das andere Gen die γ-Untereinheit. Bei der autosomal rezessiv vererbten I-Zellkrankheit ist das GNPTA-Gen (GNPTAB) für die α- und β-Untereinheit auf Chromosom 12 Genlocus q23.3 mutiert.[9] Das Gen für die γ-Untereinheit (GNPTG) ist dagegen auf Chromosom 16 Genlocus p13.3 und von der Mutation nicht betroffen.[10]
Therapie
Eine Heilung ist bisher ausgeschlossen. Die Behandlung erfolgt rein symptomatisch.
Prognose
Die Prognose ist ausgesprochen ungünstig. Die Betroffenen überleben meist nicht die erste Lebensdekade. Todesursache sind meist rezidivierende Pneumonien.[5]
Einzelnachweise
- ↑ J. G. Leroy, R. I. De Mars: Mutant enzymatic and cytological phenotypes in cultured human fibroblasts. In: Science, 157/1967, S.804-6.
- ↑ Gesellschaft für Mukopolysaccharidosen: Mukolipidose II/III: Diagnose, abgerufen am 26. März 2008
- ↑ A. Hasilik, K. von Figura: Oligosaccharides in lysosomal enzymes. Distribution of high-mannose and complex oligosaccharides in cathepsin D and b-hexosaminidase. In: Eur. J. Biochem. 121, 1981, S. 125–129.
- ↑ a b C. Schwab: Transport und Modifizierung lysosomaler Enzyme in Mukolipidose-II/III-Zellen, Dissertation, Philipps-Universität Marburg, Medizin, 1998.
- ↑ a b S. Kornfeld, W. S. Sly: I-cell disease and pseudo-Hurler polydystrophy: Disorders of lysosomal enzyme phosphorylation and localization. In: The Metabolic and Molecular Bases of Inherited Disease, Vol.II, McGraw-Hill, New York, 1995, S.2495-508.
- ↑ V. Hieber u. a.: Accumulation of [35]-S-mucopolysaccharides in cultured mucolipidosis cells. in: Birth Defects, 6/1975, S.307-10.
- ↑ G. Dawson u. a.: Glycosphingolipids in cultured human skin fibroblasts. Characterization and metabolism in fibroblasts from patients with inborn errors of glycosphingolipid and mucopolysaccharide metabolism. In: J. Biol. Chem. 247/1972, S.5951-85.
- ↑ G. H. Thomas u. a.: Increased levels of sialic acid associated with sialidase deficiency in I-cell disease fibroblats. In: Biochem. Biophys. Res. Commun., 71/1976, S.188-95.
- ↑ S. Tiede u. a.: Mukolipidosen In: Monatsschrift Kinderheilkunde, 154/2006, S.955-61.
- ↑ S. Tiede u. a.: Mucolipidosis II is caused by mutations in GNPTA encoding the alpha/beta GlcNAc-1-phosphotransferase. In: Nat. Med., 11/2005, S.1109-12. PMID 16200072 doi=10.1038/nm1305
Literatur
- C. Schwab: Transport und Modifizierung lysosomaler Enzyme in Mukolipidose-II/III-Zellen, Dissertation, Philipps-Universität Marburg, Medizin, 1998.
- J. G. Leroy u. a.: I-cell disease: biochemical studies. In: Pediatr. Res., 6/1972, S.752-7.
- M. Pavelka, J. Roth: Funktionelle Ultrastruktur - Atlas der Biologie und Pathologie von Geweben., Springer, 2005, S.100-101. ISBN 978-3-211-83563-0
- J. C. Navarro: Pathologie des Nervensystems V., Springer, 1991, S.145-148. ISBN 3-540-52873-3
- S. Tiede: Synthese des Mannose-6-Phosphat-Erkennungsmarkers lysosomaler Enzyme: Molekulare Analyse der humanen UDP-N-Acetylglucosamin:lysosomales Enzym-N-Acetylglucosamin-1-Phosphotransferase., Dissertation, Universität Hamburg, 2005.
Weblinks

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Lysosomale Speicherkrankheit
- Krankheitsbild in der Kinderheilkunde
- Zellbiologie
Wikimedia Foundation.