- Morbus Tay-Sachs
-
Klassifikation nach ICD-10 E75.0 GM2-Gangliosidose
Tay-Sachs-KrankheitICD-10 online (WHO-Version 2006) Das Tay-Sachs-Syndrom, auch unter den Bezeichnungen Morbus Tay-Sachs und infantile amaurotische Idiotie (angeborene schwerste Intelligenzminderung mit Erblindung) bekannt, ist eine autosomal-rezessiv vererbte, mit Morbus Sandhoff zu den GM2-Gangliosidosen mit Hexosaminidasedefekt gehörende Fettstoffwechselstörung. Sie ist nach dem britischen Augenarzt Warren Tay (* 1843, † 15. Mai 1927) und dem US-amerikanischen Neurologen Bernard Sachs (* 2. Januar 1858, † 8. Februar 1944) benannt, welche die Krankheit erstmals in den Jahren 1881 bzw. 1898 dokumentierten. Die Krankheit führt zu progressiver Reduktion kognitiver Fähigkeiten, psychomotorischem Abbau, muskulärer Hypotonie, Lähmung, Spastik, Blind- und Taubheit, Krämpfen, zum kirschroten Fleck in der Makula und innerhalb weniger Jahre zum Tode.
Inhaltsverzeichnis
Epidemiologie
Die Häufigkeit der für die Krankheit verantwortlichen Mutation (Chromosom 15, Lokus 15q23-24, auch Alphakette genannt) ist bei Menschen jüdisch-osteuropäischer Herkunft auffällig erhöht. Sie kommt auch besonders häufig bei Franko-Kanadiern, Iren und Cajuns vor [1]. Dort wird die Häufigkeit der heterozygoten Anlageträger mit 1:25 eingeschätzt.
Das Auftreten der Krankheit wird durch genetische Untersuchungen im Vorfeld von Schwangerschaften und bei entsprechendem Befund mit dem Ziel von deren Vermeidung niedrig gehalten.
Ätiologie
GM2-Ganglioside werden normalerweise kontinuierlich durch sequentielle Abspaltung der endständigen Zucker abgebaut. Den betroffenen Kindern fehlt das Enzym β-N-Acetylhexosaminidase, das für die Entfernung von terminalen N-Acetylgalactosaminreste zuständig ist. Daher ist der Gangliosidgehalt in Gehirn und Retina des Kindes drastisch erhöht. Nach Aufblähung der befallenen Nervenzellen kommt es schließlich zu deren Untergang.
Diagnose
Die Krankheit wird meistens zwischen dem dritten und achten Lebensmonat erkannt. Der Nachweis ist aufgrund verminderter Aktivität von Hexosaminidase A bzw. B in Blutserum, Leukozyten- oder Fibroblastenkulturen gegeben. Auch ein Heterozygotennachweis ist möglich (Pränataldiagnostik).
Symptomatik
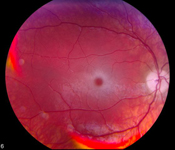 Veränderungen der Netzhaut, der rote Fleck innerhalb der Makula
Veränderungen der Netzhaut, der rote Fleck innerhalb der Makula- Kirschroter Fleck auf der Makula bei über 95% der Patienten
- Zunehmende Muskelschwäche nach dem dritten Lebensmonat
- Schreckreaktionen auf Schallreize
- Psychomotorischer Abbau, Verlust des Sitz- und Stehvermögens
- Zunehmende Schwerhörigkeit, Blindheit, Paresen sowie Spasmen
- Puppenartiges Gesicht mit blasser durchscheinender Haut, langen Augenwimpern, feinem Haar und auffällig rosafarbener Gesichtsfarbe
Ergänzend hierzu:
- In der späten Säuglingszeit zunehmendes Erbrechen sowie rezidivierende Pneumonien
- Nach dem 16. Lebensmonat progressive Makrozephalie infolge zerebraler Gliose. Zudem tritt die Lipidose kortikaler, autonomer und rektaler Mukosaneuronen mit balloniertem Zytoplasma und peripher abgedrängtem Zellkern auf. Fortschreitende Demyelinisierung sowie kortikale Gliose sind ein weiteres Merkmal. Hierbei sind keine pathologischen Veränderungen viszeraler Organe zu beobachten.
Prognose
Die Patienten versterben in der Regel bis zum dritten Lebensjahr aufgrund einer rezidivierenden Pneumonie.
Therapie
Es können lediglich die Symptome therapiert werden.
In besonders betroffenen Bevölkerungsgruppen werden zur Erfassung heterozygoter Anlageträger entsprechende Beobachtungsprogramme durchgeführt. Familien in denen die Krankheit bereits aufgetreten ist, nutzen die Möglichkeit einer genetischen Beratung im Vorfeld einer Schwangerschaft bzw. die pränatale Diagnostik. Um die Krankheit zu vermeiden, ist von einer Schwangerschaft abzuraten.
Einzelnachweise

Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.