- Polymerasekettenreaktion
-
Die Polymerase-Kettenreaktion (englisch Polymerase Chain Reaction, PCR) ist eine Methode, um die Erbsubstanz DNA in vitro zu vervielfältigen. Dazu wird ein Enzym verwendet, die DNA-Polymerase. Der Begriff "Kettenreaktion" beschreibt in diesem Zusammenhang die Tatsache, dass die Produkte vorheriger Zyklen als Ausgangsstoffe für den nächsten Zyklus dienen und somit eine exponentielle Vervielfältigung ermöglichen.
Die PCR wird in biologischen und medizinischen Laboratorien für eine Vielzahl verschiedener Aufgaben verwendet, zum Beispiel für die Erkennung von Erbkrankheiten und Virusinfektionen, für das Erstellen und Überprüfen genetischer Fingerabdrücke, für das Klonieren von Genen und für Abstammungsgutachten. Die PCR zählt zu den wichtigsten Methoden der modernen Molekularbiologie und viele wissenschaftliche Fortschritte auf diesem Gebiet (z. B. im Rahmen des Humangenomprojekts) wären ohne diese Methode nicht denkbar gewesen.
Inhaltsverzeichnis
Geschichte
Anfang der 1970er Jahre kam der norwegische Postdoc Kjell Kleppe im Labor von Nobelpreisträger Har Gobind Khorana auf den Gedanken, DNA durch zwei flankierende Primer zu vervielfältigen, jedoch geriet die Idee in Vergessenheit.[1] Die Polymerase-Kettenreaktion selber wurde 1983 von Kary Mullis erneut erfunden. Seine Absicht war es, ein neuartiges DNA-Syntheseverfahren zu entwickeln, das DNA durch wiederholte Verdopplung in mehreren Zyklen mit Hilfe eines Enzyms namens DNA-Polymerase künstlich vervielfältigt. Sieben Jahre, nachdem er seine Idee veröffentlicht hatte, wurde Mullis hierfür 1993 der Nobelpreis für Chemie verliehen.[2]
DNA-Polymerase kommt in allen Lebewesen vor und verdoppelt während der Replikation die DNA vor der Zellteilung. Dazu bindet sie sich an einen einzelnen DNA-Strang und synthetisiert mit Hilfe eines kurzen komplementären Oligonukleotids (Primer) einen dazu komplementären Strang. Bereits in Mullis' ursprünglichem PCR-Versuch wurde das Enzym in vitro verwendet. Die doppelsträngige DNA wurde zunächst durch Erhitzen auf 96 ° C in zwei Einzelstränge getrennt. Bei dieser Temperatur wurde die dabei zunächst verwendete DNA-Polymerase I von E. coli zerstört und musste daher nach jedem Erhitzen erneut zugegeben werden. Diese Folge von Arbeitsschritten musste mehrere dutzendmal in Folge wiederholt werden, um eine ausreichende Amplifikation zu erreichen. Mullis’ ursprüngliches Verfahren war daher sehr ineffizient, da es viel Zeit, große Mengen DNA-Polymerase und ständige Aufmerksamkeit erforderte.
Eine entscheidende Verbesserung der PCR-Technologie brachte die Verwendung von thermostabilen DNA-Polymerasen, d. h. Enzymen, die auch bei Temperaturen von annähernd 100 °C ihre Polymerase-Aktivität behalten. Eine der ersten thermostabilen DNA-Polymerasen wurde aus dem in heißen Quellen lebenden thermophilen Bakterium Thermus aquaticus gewonnen und Taq-Polymerase genannt. Durch die Verwendung thermostabiler DNA-Polymerasen bestand keine Notwendigkeit mehr, ständig neue Polymerase zuzugegeben, und der ganze PCR-Prozess konnte erheblich vereinfacht und automatisiert werden.
Mullis arbeitete zu dieser Zeit für die kalifornische Biotech-Firma Cetus und wurde mit einer Prämie von 10.000 US-Dollar abgefunden. Jahre später verkaufte Cetus dann die Rechte an der PCR-Methode mitsamt dem Patent für die von ihm verwendete DNA-Polymerase Taq an die Firma Roche für 300 Millionen Dollar. Das Enzym Taq wurde bereits 1980 von dem russischen Forscher Kaledin beschrieben. Aus diesem Grund wurde nach jahrelangem Rechtsstreit der Firma Roche das Patent für Taq inzwischen entzogen. Die US-Patente für die PCR-Technologie selber liefen im März 2005 aus.
Die Taq-Polymerase erfährt nach wie vor breite Anwendung. Ihr Nachteil liegt darin, dass sie manchmal Fehler beim Kopieren der DNA produziert, was zu Mutationen in der DNA-Sequenz führt. Polymerasen wie Pwo und Pfu, die aus Archaea gewonnen werden, haben einen Korrektur-Mechanismus, der die Anzahl der Mutationen in der kopierten DNA erheblich senkt.
 Zwei Thermocycler mit einem gemeinsamen Bedienelement
Zwei Thermocycler mit einem gemeinsamen Bedienelement geöffneter Thermocycler, Heizblock mit 8 PCR-Reaktionsgefäßen
geöffneter Thermocycler, Heizblock mit 8 PCR-ReaktionsgefäßenPCR in der Praxis
PCR wird eingesetzt, um einen kurzen, genau definierten Teil eines DNA-Strangs zu vervielfältigen. Dabei kann es sich um ein Gen oder auch nur um einen Teil eines Gens handeln oder auch um nicht-kodierende DNA-Sequenzen. Im Gegensatz zu lebenden Organismen kann der PCR-Prozess nur relativ kurze DNA-Abschnitte kopieren. Bei einer Standard-PCR können dies bis zu etwa 3.000 Basenpaare (3 kbp) lange DNA-Fragmente sein. Mit Hilfe bestimmter Polymerasen-Gemische, weiterer Additive in der PCR und optimalen Bedingungen können sogar Fragmente mit einer Länge von über 20–40 kbp vervielfältigt werden, was immer noch sehr viel kürzer ist als die chromosomale DNA einer eukaryotischen Zelle. Eine menschliche Zelle enthält beispielsweise etwa drei Milliarden Basenpaare pro haploidem Genom.
In ihren momentanen Anwendungsgebieten benötigt PCR mehrere grundlegende Komponenten. Diese sind:
- Die Original-DNA, die den zu vervielfältigenden Abschnitt enthält (Template)
- Zwei Primer, um auf den beiden Einzelsträngen der DNA jeweils den Startpunkt der DNA-Synthese festzulegen, wodurch der zu vervielfältigende Bereich von beiden Seiten begrenzt wird.
- DNA-Polymerase, die bei hohen Temperaturen nicht zerstört wird, um den festgelegten Abschnitt zu replizieren (kopieren) (z. B. Taq-Polymerase)
- Desoxyribonucleosidtriphosphat, die Bausteine für den von der DNA-Polymerase synthetisierten DNA-Strang
- Mg2+-Ionen, für die Funktion der Polymerase essentiell
- Pufferlösungen, die eine für die DNA-Polymerase geeignete chemische Umgebung sicherstellen
Die Polymerase-Kettenreaktion findet in einem sogenannten Thermocycler statt. Diese Maschine erhitzt und kühlt die in ihr befindlichen Reaktionsgefäße präzise auf die Temperatur, die für den jeweiligen Schritt benötigt wird. Um Verdunstung zu verhindern, wird ein dicht schließendes Reaktionsgefäß oder eine Ölschicht auf dem Reaktionsgemisch verwendet. Etwaige Kondensatbildung im Deckel des Gefäßes wird durch einen beheizbaren Gerätedeckel (über 100 °C) verhindert.
Soll die PCR vor allem als quantitativer Nachweis dienen, empfiehlt sich die sog. Real Time PCR.
Die Verwendung einer Pyrophosphatase kann unter Umständen die Effektivität der PCR steigern. Das Enzym katalysiert die Hydrolyse des von den Nukleotidtriphosphaten abgespaltenen Pyrophosphats zu Orthophosphat. Pyrophosphat kann als Inhibitor bei der PCR wirken.
 PCR-Reaktionsgefäße.
PCR-Reaktionsgefäße. PCR-Platte für 96 Ansätze mit Gummiabdeckung
PCR-Platte für 96 Ansätze mit GummiabdeckungBeispiel
Als allgemeines Beispiel sei hier die „Rezeptur“ für eine PCR-Reaktion wiedergegeben (viele Beispiele für die verschiedensten Reaktionen finden sich ansonsten in der wissenschaftlichen Literatur in allen möglichen Variationen):
-
1,0 µl DNA-Lösung (100 ng/µl) 2,0 µl pro Primer(i.d.R. zwei) (10 µM) 1,0 µl Pfu-, Taq- oder andere thermostabile Polymerase (1–5 U/µl) 1,0 µl 10 mM Desoxy-Nukleotide (dATP, dGTP, dCTP, dTTP), „dNTP“ 5,0 µl 10-fach konzentrierte Polymerase-Pufferlösung 38,0 µl H2O 50,0 µl Gesamtvolumen
Ein 200-µl-Reaktionsgefäß mit den 50 µl Gemisch wird in den Thermocycler gestellt. Als Reaktionsgefäß können neben einzelnen 200-µl-Reaktionsgefäßen auch acht zusammenhängende 200-µl-Reaktionsgefäße oder PCR-Platten für bis zu 96 Ansätze verwendet werden. Die Platten werden entweder mit einer Gummiabdeckung oder einer selbstklebenden Klarsichtfolie verschlossen.
Theoretischer Ablauf
Der PCR-Prozess besteht aus einer Anzahl von 12–50 Zyklen, die in einem Thermocycler durchgeführt werden. Die folgenden Angaben sind als Richtwerte gedacht. Meist muss eine PCR auf die spezifische Reaktion hin optimiert werden. Jeder Zyklus besteht aus drei Schritten (siehe Abbildung unterhalb):
- Denaturierung (Melting, Schmelzen): Zunächst wird die doppelsträngige DNA auf 94–96 °C erhitzt, um die Stränge zu trennen. Die Wasserstoffbrückenbindungen, die die beiden DNA-Stränge zusammenhalten, werden aufgebrochen. Im ersten Zyklus wird die DNA oft für längere Zeit erhitzt (Initialisierung), um sicherzustellen, dass sich sowohl die Ausgangs-DNA als auch die Primer vollständig voneinander getrennt haben und nur noch Einzelstränge vorliegen. Manche (so genannte hot start-) Polymerasen müssen durch eine noch längere anfängliche Erhitzungs-Phase (bis zu 15 Minuten) aktiviert werden.
- Primerhybridisierung (primer annealing): Die Temperatur wird ca. 30 Sekunden lang auf einer Temperatur gehalten, die eine spezifische Anlagerung der Primer an die DNA erlaubt. Die genaue Temperatur wird hierbei durch die Länge und die Sequenz der Primer bestimmt (bzw. der passenden Nukleotide im Primer, wenn durch diesen Mutationen eingeführt werden sollen = site-directed Mutagenesis). Wird die Temperatur zu niedrig gewählt, können sich die Primer unter Umständen auch an nicht-100-%-komplementären Sequenzen anlagern und so zu unspezifischen Produkten („Geisterbanden“) führen. Wird die Temperatur zu hoch gewählt, ist die thermische Bewegung der Primer u. U. so groß, dass sie sich nicht richtig anheften können, so dass es zu gar keiner oder nur ineffizienter Produktbildung kommt. Die Temperatur, welche die beiden oben genannten Effekte weitgehend ausschließt, liegt normalerweise 2–3 °C unter dem Schmelzpunkt der Primersequenzen; dies entspricht meist einer Temperatur von 55–65 °C.
- Elongation (Polymerisation, Verlängerung, Amplifikation): Schließlich füllt die DNA-Polymerase die fehlenden Stränge mit freien Nukleotiden auf. Sie beginnt am 3'-Ende des angelagerten Primers und folgt dann dem DNA-Strang. Der Primer wird nicht wieder abgelöst, er bildet den Anfang des neuen Einzelstrangs. Die Temperatur hängt vom Arbeitsoptimum der verwendeten DNA-Polymerase ab (68–72 °C). Dieser Schritt dauert etwa 30 Sekunden je 500 Basenpaare, variiert aber in Abhängigkeit von der verwendeten DNA-Polymerase. Übliche Thermocycler kühlen die Reaktionsansätze nach Vollendung aller Zyklen auf 4–8 °C, so dass eine PCR am Abend angesetzt werden kann und die Proben am Morgen darauf weiter verarbeitet werden können.
 Schematische Darstellung des PCR-Zyklus.
Schematische Darstellung des PCR-Zyklus.
(1) Schmelzen (Denaturierung) bei ca. 96 °C.
(2) Anlagerung (Primerhybridisierung) bei ca. 68 °C.
(3) Verlängerung (Elongation) bei ca. 72 °C (P=Polymerase).
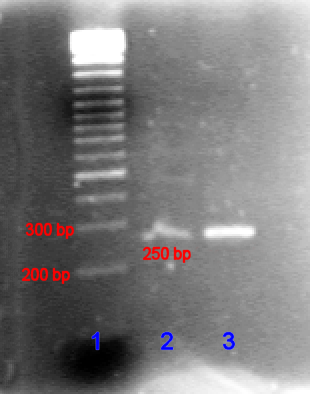
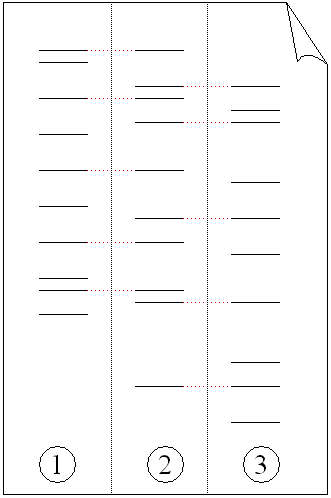
Exponentielles Anwachsen des Kurzen Produktes (von Primern eingeschlossener Bereich). Das PCR-Produkt in niedriger (Bande 2) und hoher (Bande 3) Konzentration im Vergleich mit der DNA-Leiter (Bande 1) in Agarose-Gel.
Das PCR-Produkt in niedriger (Bande 2) und hoher (Bande 3) Konzentration im Vergleich mit der DNA-Leiter (Bande 1) in Agarose-Gel.Im ersten Zyklus entstehen pro DNA-Ausgangsdoppelstrang 2 DNA-Stränge, welche im Bereich der Zielsequenz doppelsträngig sind. Nach dem Schmelzen am Anfang des zweiten Zyklus stehen dadurch die beiden ursprünglichen DNA-Einzelstränge und zwei am 3’-Ende überlange Einzelstränge zur Verfügung. Dies ist damit zu erklären, dass lediglich ein Startpunkt (Primer), nicht aber ein Endpunkt exakt festgelegt ist. Der Abbruch der Strangsynthese erfolgt dabei spätestens durch die Strangtrennung im folgenden Denaturierungsschritt. Im zweiten Zyklus stehen die eingesetzte DNA sowie die gerade gebildeten DNA-Stränge zur Verfügung. An Ersterer erfolgt derselbe Prozess wie im ersten Zyklus. An die neu gebildeten DNA-Einzelstränge, welche an 3’ bereits dort enden wo sie sollen, lagern sich nun Primer in der 5’-Region an. Die nun gebildeten Stränge haben keinen 5’-Überhang, da die Polymerase auf dem Template in Richtung des 3’-Endes liest. Am Ende des zweiten Zyklus stehen damit erstmals Produkte der gewünschten Länge zur Verfügung. In den folgenden Zyklen vermehren sich die gewünschten Produkte exponentiell (da sie selbst als Matrize für weitere Strangsynthesen dienen), während die ungewünschten langen Produkte (siehe Produkte des ersten Zyklus) nur linear ansteigen (nur eingesetzte DNA dient als Matrize). Dies ist der theoretische Idealfall, in der Praxis fallen zudem in geringem Maße auch kürzere Fragmente als die gewünschte Ziel-DNA an. Diese kurzen Fragmente häufen sich vor allem in den späten Zyklen an, wodurch meist nur etwa 30 Zyklen durchlaufen werden, um insgesamt vorwiegend DNA der gewünschten Länge und Sequenz zu erhalten.
Das PCR-Produkt kann durch Agarose-Gelelektrophorese anhand seiner Größe identifiziert werden. (Die Agarose-Gelelektrophorese ist ein Verfahren, bei der DNA in ein Agarose-Gel eingebracht wird und anschließend eine Spannung angelegt wird. Dann bewegen sich die kürzeren DNA-Stränge schneller als die längeren auf den Pluspol zu.) Die Länge des PCR-Produkts kann durch einen Vergleich mit einer DNA-Leiter, die DNA-Fragmente bekannter Größe enthält und parallel zur Probe im Gel mitläuft, bestimmt werden.
PCR-Anwendungsgebiete
Die PCR kann für eine Vielzahl von Experimenten und Analysen eingesetzt werden, einige Beispiele werden weiter unten vorgestellt.
Genetischer Fingerabdruck
Der genetische Fingerabdruck wird in der Gerichtsmedizin zur Identifizierung einer Person eingesetzt, indem ihre DNA mit einer vorhandenen Probe verglichen wird. Beispielsweise kann eine Blutprobe von einem Tatort mit dem Blut eines Verdächtigen verglichen werden. Die Probe kann minimal sein, das heißt, eine einzige Zelle (nur weiße Blutzellen kommen in Frage, da in roten kein Kern ist) genügt. An Tatorten findet man meistens Blut, Sperma, Speichel, Haut, Haare oder ähnliche Zellen (Speichel und Haare enthalten zwar keine Zellkerne, jedoch sind im Speichel meist Zellreste zu finden, an Haaren oft die Haarwurzeln). Theoretisch genügt ein einziger Strang. Zuerst spaltet man die DNA-Probe in Fragmente auf, die dann mittels PCR vervielfältigt werden. Die vervielfältigten Fragmente werden anschließend durch Gelelektrophorese getrennt. Die so gewonnene Anordnung der DNA-Fragmente nennt man DNA-Fingerabdruck. Diese Fragmente enthalten jedoch größtenteils polymorphe Bereiche. Das sind sich wiederholende DNA-Abschnitte im nicht kodierenden Bereich der DNA (junk DNA), sogenannte repetetive Sequenzen. Diese Sequenzen liegen zwischen den Genen. Deshalb lässt sich anhand des genetischen Fingerabdrucks keine Disposition feststellen.
Vaterschaftstest
 Elektrophorese von PCR vervielfältigter DNA-Fragmente. (1) Vater. (2) Kind. (3) Mutter. Das Kind hat Teile der Fingerabdrücke der beiden Elternteile geerbt wodurch es über einen eigenen, einzigartigen Fingerabdruck verfügt.
Elektrophorese von PCR vervielfältigter DNA-Fragmente. (1) Vater. (2) Kind. (3) Mutter. Das Kind hat Teile der Fingerabdrücke der beiden Elternteile geerbt wodurch es über einen eigenen, einzigartigen Fingerabdruck verfügt.Obwohl diese erzeugten „Fingerabdrücke“ einzigartig sind (bei eineiigen Zwillingen sind sie nahezu identisch; Unterschiede können hier nur mit zusätzlichem Aufwand nachgewiesen werden), können genetische Beziehungen, zum Beispiel zwischen Eltern und Kindern oder zwischen Geschwistern, durch zwei oder mehr genetische Fingerabdrücke bestimmt werden, was bei Vaterschaftstests zum Einsatz kommt (siehe Abbildung). Eine Abwandlung dieser Technik wird auch zur Bestimmung evolutionärer Beziehungen zwischen Organismen angewandt.
Erkennung von Krankheiten
Die Erkennung von Erbkrankheiten in einem vorliegenden Genom ist ein langwieriger und komplizierter Vorgang, der durch den Einsatz von PCR bedeutend verkürzt werden kann. Jedes Gen, das in Frage kommt, kann durch PCR mit den entsprechenden Primern amplifiziert (= vervielfältigt) und anschließend sequenziert werden (DNA sequenzieren heißt, die Sequenz der Nukleotide (oder Basen) der DNA zu bestimmen), um Mutationen aufzuspüren.
Virale Erkrankungen können ebenfalls durch PCR erkannt werden, indem man die Virus-DNA vervielfältigt bzw. bei RNA-Viren diese RNA erst in DNA umschreibt und dann mittels PCR vervielfältigt (die RT-PCR). Diese Analyse kann sofort nach der Infektion erfolgen, oft Tage oder Wochen vor dem Auftreten der Symptome. Erfolgt die Diagnose so früh, erleichtert das den Medizinern die Behandlung erheblich.
Die PCR kann auch zu Reihenuntersuchungen eingesetzt werden. So wird sie z. B. von Blutspendediensten zur Routineuntersuchung von Blutkonserven eingesetzt. Durch die frühestmögliche Entdeckung einer gefährlichen Infektionskrankheit beim Spender (z. B. HIV, Hepatitis B) kann das sog. diagnostische Fenster nahezu geschlossen werden. Durch die Empfindlichkeit der PCR-Tests ist es möglich, Proben in sog. Pools (z. B. 96 Einzelproben) zusammenzufassen. Wird ein Pool positiv getestet, wird seine Größe solange verkleinert (meistens halbiert), bis die verursachende Probe gefunden ist.
Klonierung von Genen
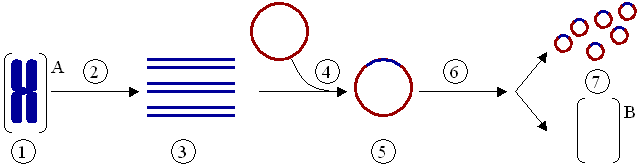
Das Klonieren eines Gens – nicht zu verwechseln mit dem Klonen eines ganzen Organismus – ist ein Vorgang, bei dem ein Gen aus einem Organismus isoliert und anschließend in einen anderen eingepflanzt wird. PCR wird oft benutzt, um das Gen zu vervielfältigen, das dann in einen Vektor (ein Vektor ist ein Mittel, mit dem ein Gen in einen Organismus verpflanzt werden kann), beispielsweise ein Plasmid (ein ringförmiges DNA-Molekül), eingefügt wird (Abb. 4). Die DNA kann anschließend in einen anderen Organismus eingesetzt werden, in dem das Gen oder sein Produkt besser untersucht werden kann. Das Exprimieren eines klonierten Gens kann auch zur massenhaften Herstellung nutzbarer Proteine wie z. B. Arzneimittel dienen.
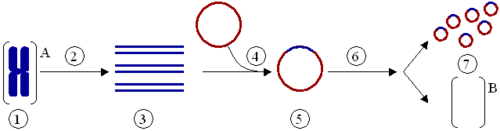 Klonierung eines Gens mit Hilfe eines Plasmids:
Klonierung eines Gens mit Hilfe eines Plasmids:
(1) Chromosomale DNA von Organismus A.
(2) PCR.
(3) Mehrere Kopien eines einzelnen Gens von Organismus A.
(4) Einfügen des Gens in ein Plasmid.
(5) Plasmid mit dem Gen aus Organismus A.
(6) Einfügen des Plasmids in Organismus B.
(7) Vervielfältigung oder Expression des Gens, das aus Organismus A stammt, im Organismus B.Mutagenese
Mutagenese ist eine Möglichkeit, die Sequenz der Nukleotide (Basen) der DNA zu verändern. Es gibt Situationen, in denen man mutierte (veränderte) Kopien eines bestimmten DNA-Strangs benötigt, um die Funktion eines Gens zu bestimmen. Mutationen können in kopierte DNA-Sequenzen auf zwei grundsätzlich verschiedene Arten während des PCR-Prozesses eingefügt werden.
Gezielte Mutagenese (z. B.: “site-directed mutagenesis”) erlaubt es dem Forscher, an spezifischen Stellen auf dem DNA-Strang Mutationen zu erzeugen. Meist wird dafür die gewünschte Mutation in die Primer integriert, die für die PCR verwendet werden. Bei der gezielten bzw. stellenspezifischen Mutagenese ist mindestens einer der Primer nicht 100prozentig identisch mit der DNA, an die er sich anlagert. Während der Amplifikation wird so eine Mutation in das DNA-Fragment eingeführt.
Zufällige Mutagenese (“random mutagenesis”) beruht hingegen auf der Verwendung von fehlerträchtigen Polymerasen (bzw. Polymerasen ohne Mechanismus zur Fehlerkorrektur) während des PCR-Prozess. Bei der zufälligen Mutagenese können Ort und Art der Mutationen nicht beeinflusst werden und müssen erst durch eine Sequenzierung identifiziert werden.
Eine Anwendung der zufälligen oder gezielten Mutagenese ist die Analyse der Struktur-Funktions-Beziehungen eines Proteins. Nach der Veränderung der DNA-Sequenz kann man das entstandene Protein mit dem Original vergleichen und die Funktion aller Teile des Proteins bestimmen. Weiterhin können damit auch Funktionen der Nukleinsäure selbst (mRNA-Transport, mRNA-Lokalisation, etc.) untersucht werden.
Analyse alter (fossiler) DNA
Da die PCR aus nur geringen DNA-Probemengen eine beliebige Menge von Material erzeugen kann, ist sie besonders für die sehr alte aDNA geeignet, die in der Natur nur noch in für Untersuchungen nicht mehr ausreichenden Mengen vorkommt. Dabei beruhen nahezu alle wissenschaftlichen Erkenntnisgewinne in Bezug auf die aDNA und somit viele seit langem ausgestorbener Arten auf der Methode der PCR.
Geschlechtsbestimmung
Die PCR kann eingesetzt werden, um Material für einen Nachweis der geschlechtsspezifischen Chromosomen zu gewinnen. Das ist bei den üblichen Haussäugetieren nicht üblich, außer eventuell bei Zwittern. Bei manchen Tieren im Zoo, besonders aber bei Vögeln, Reptilien oder Fischen, ist es die schonendste und sicherste Variante. Im Heimtierbereich ist diese PCR Methode bei Papageien üblich.
Siehe auch
- Sequenzanalyse
- Elektrophorese
- Ligase-Kettenreaktion
- Reverse-Transkriptase-Polymerasekettenreaktion
- Real time quantitative PCR
- Inverse Polymerase-Kettenreaktion
- Immuno-Polymerasekettenreaktion
Quellen
- ↑ Kleppe, K. et al. (1971): Studies on polynucleotides. XCVI. Repair replications of short synthetic DNAs as catalyzed by DNA polymerases. In: J. Mol. Biol. Bd. 56, S. 341–361. PMID 4927950
- ↑ Informationen der Nobelstiftung zur Preisverleihung 1993 an Kary B. Mullis (englisch)
Literatur
- Zur PCR (Polymerase-Kettenreaktion)
- C. R. Newton, A. Graham: PCR. Introduction to Scientific Techniques. 2d. ed. BIOS Scientific Publishers Ltd., Oxford 1997. ISBN 1-872748-82-1
- R. K. Saiki, D. H. Gelfand, S. Stoffel, S. J. Scharf, R. Higuchi, G. T. Horn, K. B. Mullis, H. A. Erlich. Primer-Directed Enzymatic Amplification of DNA with a Thermostable DNA Polymerase. in: Science. 239.1988, 487–491. ISSN 0036-8075 PMID: 2448875
- Mullis: System for automated performance of the polymerase chain reaction. United States Patent 5,656,493, August 12, 1997.
- Rabinow, Paul: Making PCR: A Story of Biotechnology, University of Chicago Press, 1996. ISBN 0-226-70146-8
- Zum Genetischen Fingerabdruck (Genetic Fingerprinting)
- J. Ballantyne (Hrsg.): DNA Technology and Forensic Science. Cold Spring Harbor Laboratory, Cold Spring Harbor NY 1989. ISBN 0-87969-232-4
- P. R. Billings (Hrsg.): DNA on Trial. Genetic Identification and Criminal Justice. Cold Spring Harbor Laboratory, Cold Spring Harbor NY 1992. ISBN 0-87969-379-7
- R. K. Saiki, S. Scharf, F. Faloona, K. B. Mullis, G. T. Horn, H. A. Erlich, N. Arnheim: Enzymatic Amplification of Beta-Globin Genomic Sequences and Restriction Site Analysis for Diagnosis of Sickle-Cell Anemia. in: Science. 230.1985, 1350–1354. ISSN 0036-8075
Weblinks
- Animation der PCR mit Beschreibung (deutsch)
- Animation PCR(Flash)
- Animation zum Prinzip der PCR
- PCR Webseite zu allen Themen der PCR
- The reference in real-time PCR Webseite zu allen praktischen Aspekten der real-time PCR und real-time RT-PCR (englisch)
- Making PCR (Polymerase Chain Reaction) PCR-Projekt an der Universität von Kalifornien, Berkeley (englisch)
- PCR Anleitung (englisch)
- The World Famous PCR Jump Station: Polymerase Chain Reaction Internetseite zu allen praktischen Aspekten der PCR (englisch)
- englischer Internet-Link zur optimalen Berechnung der annealing-Temperatur von Primern
- Primerfox - kostenloses Tool zur Erstellung passender PCR Primer.
- Der PCR Song Scientists for better PCR




Wikimedia Foundation.