- Zystennieren
-
Klassifikation nach ICD-10 Q61.1 Polyzystische Niere, autosomal-rezessiv
infantiler TypQ61.2 Polyzystische Niere, autosomal-dominant
ErwachsenentypQ61.3 Polyzystische Niere, nicht näher bezeichnet ICD-10 online (WHO-Version 2006) 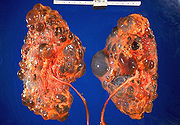 Präparat von polyzystischen Nieren
Präparat von polyzystischen Nieren Im Vergleich dazu gesunde Nieren (vom Lamm)
Im Vergleich dazu gesunde Nieren (vom Lamm)Zystennieren, auch als polyzystische Nieren bezeichnet, (engl.: polycystic kidney disease, PKD) sind eine Gruppe ernsthafter, meist erblich bedingter, Erkrankungen der Nieren.[1] Durch die Bildung einer Vielzahl (griech.: poly = πολύς = „viel“) von flüssigkeitsgefüllten Kammern beziehungsweise Bläschen, den sogenannten Zysten, sind die Nieren in ihrer Filterfunktion erheblich eingeschränkt. Bei einer Nierenzyste handelt es sich demgegenüber um eine einzelne Zyste, die im Rahmen einer Untersuchung als in aller Regel harmloser Zufallsbefund erwähnt wird.
Genetisch bedingte Zystennieren sind die häufigste lebensbedrohliche Erbkrankheit beim Menschen und eine der Hauptursachen für chronisches Nierenversagen. Eine Heilung ist nur durch eine Nierentransplantation möglich.
Symptome
Erste Symptome, die auf Zystennieren hinweisen können, sind Bluthochdruck, blutiger Urin (Hämaturie), wiederholte Harnwegsinfekte, eine Zunahme des Bauchumfangs und Schmerzen im Bauchraum. Etwa ein Drittel der Patienten bleibt, selbst bis zum Zeitpunkt des terminalen (endgültigen) Nierenversagens (end-stage renal failure, ESRF), ohne Symptome. Dies erschwert eine frühzeitige Diagnose erheblich.[2][3] Die von polyzystischen Nieren betroffenen Patienten klagen häufig über Schmerzen in der seitlichen Flanke des Rückens oder des Bauches. Die Schmerzen können dabei vorübergehend oder dauerhaft dumpf und quälend sein. Der Schmerz ist vermutlich durch das ausgedehnte Zystenwachstum bedingt. Zudem werden umgebende Organe durch die extreme Dehnung der Nierenkapsel (Capsula fibrosa renalis) verdrängt.
Die Schmerzen können durch Punktion der Zysten, beispielsweise perkutan, das heißt durch die Haut hindurch, oder minimal-invasiv durch laparoskopische Dekortikation der Zysten, kurzfristig gelindert werden.[4][5][6][7][2] Durch die Neubildung von Zysten sind diese Maßnahmen nicht nachhaltig, so dass die entsprechenden Eingriffe wiederholt werden müssen.[8] Die Behandlung ändert zudem nichts am Verlauf der Krankheit.[2]
Bei etwa 30 bis 50 % der Patienten erfolgt die Erstdiagnose „polyzystische Nieren“ über blutigen Urin (Hämaturie). Die Ursache für die Blutungen sind meist Risse der Zysten. Die Blutungen selbst sind weitgehend ungefährlich und kommen von selbst zum Stillstand.[2] Patienten mit Zystennieren scheiden – bedingt durch die eingeschränkte Nierenfunktion – erhöhte Mengen von körpereigenen Eiweißen (Proteine) über den Urin aus. Scheidet der Körper tgl. 20 bis 200 Milligramm Albumin aus, so spricht man von einer Mikroalbuminurie. Werden noch größere Mengen an Albumin ausgeschieden, so wird dies Makroalbuminurie genannt.[9] Sind im Harn größere Proteine als Albumin nachweisbar liegt eine Proteinurie vor. Letztere ist mit Teststreifen, die in den Urin gehalten werden, einfach nachweisbar. Die Mikroalbuminurie ist deutlich schwieriger feststellbar. Protein- und Mikroalbuminurie sind ein Indiz für eine eingeschränkte Funktion der Niere. Polyzystische Nieren sind nur eine mögliche Erkrankung, die zu dieser Funktionsstörung führen können.
Eine arterielle Hypertonie („Bluthochdruck“) liegt bei 50 bis 75 % der Patienten mit polyzystischen Nieren vor. Der Blutdruck der Betroffenen ist häufig bereits vor einem Leistungsabfall der Nierenfunktion (Glomeruläre Filtrationsrate, GFR) deutlich erhöht.[10][11][12]
Diagnostik
 Zystennieren beidseits (gelb umrandet) in einer Magnetresonanztomographie
Zystennieren beidseits (gelb umrandet) in einer MagnetresonanztomographieDie Diagnose wird in der Regel durch eine Sonografie („Ultraschall“)[13] oder andere bildgebende Verfahren, wie beispielsweise der Magnetresonanztomographie,[14] gestellt.[15] Mit der Sonographie können in modernen Geräten Zysten bis herab zu einer Größe von 5 mm diagnostiziert werden. Die Früherkennungsrate bei 20-jährigen Patienten liegt bei etwa 90 %.[16] Die Computertomographie bietet zwar eine höhere Auflösung mit besserer Bildqualität, sie wird jedoch vor allem wegen der Strahlenbelastung nicht für Patienten-Screenings, sondern nur bei speziellen diagnostischen Fragestellungen eingesetzt.[2]
Die Biopsie, bei der eine kleine Menge von Nieren- aber auch Lebergewebe entnommen wird, dient im Kindesalter der Differenzierung zwischen ARPKD und ADPKD (early-onset). Damit können morphologische Veränderungen der Basalmembran schon zu einem sehr frühen Zeitpunkt nachgewiesen werden. Über die Bestimmung einer kongenitalen Leberfibrose erfolgt die Diagnose einer ARPKD.[2]
Nach Osathanondh und Potter werden die Zystennieren pathologisch-anatomisch in folgende Typen eingeteilt:[17]
Typ Befall Nierengröße Zystengröße Glomeruli Gallengangzysten Überlebenszeit I beidseitig vergrößert bis stark vergrößert gleichmäßig weit (12 mm) normal vorhanden Neugeborenenperiode II beidseitig, einseitig oder partiell vergrößert oder verkleinert unterschiedlich groß vermindert und abnormal nicht vorhanden Erwachsenenalter III meist bilateral vergrößert unterschiedlich groß, zum Teil sehr weit glomeruläre Zysten gelegentlich, dann aber nur in umschriebenen Arealen Erwachsenenalter IV beidseitig verkleinert klein, subkapsulär gelegen Glomeruli vermindert, glomuläre Zysten nicht vorhanden Erwachsenenalter Die so definierten Typen ermöglichen in der Praxis jedoch oft keine eindeutige Klassifikation. Neben der pathoanatomischen Beschreibung von Nieren und Leber spielt daher die familiäre Vorgeschichte (Anamnese) eine wichtige Rolle. Für die erblich bedingten Fälle von Nierenzysten werden daher meist die genetisch fundierten Begriffe autosomal-dominant und autosomal-rezessiv verwendet.[18]
Durch die Identifizierung von potenziell betroffenen Genen ist eine nichtinvasive beweisende molekulargenetische Diagnostik möglich. Dieses Verfahren kann in vielen Fällen die invasive Biopsie-Diagnostik ersetzen und eine ätiologische Klassifizierung ermöglichen. Diese Klassifizierung eröffnet wiederum Wege für differentialtherapeutische Möglichkeiten zur Behandlung der Erkrankung. Die Sensitivität für ein richtig positives Ergebnis liegt dabei bei ungefähr 95 %.[2] Eine Korrelation zwischen Genotyp und Phänotyp ist nur eingeschränkt möglich.[19] Die Mutationsanalytik gestaltet sich beim PKD1-Gen durch seine Größe (46 kodierende Exons und 14,2 kb des Transkripts) schwierig. Hinzu kommt, dass bei dem betroffenen Chromosom 16) auf Genlocus p13.1 die ersten 33 Exons von PKD1 in drei homologen Kopien (HG-A ≈21 kb, HG-B ≈17 kb und HG-C ≈8,5 kb; HG = homologes Gen) vorliegen. Dies erschwert die spezifische Vervielfachung mittels Polymerase-Kettenreaktion (PCR) erheblich.[20]
Aus der molekulargenetischen Diagnostik ergibt sich eine besondere Problematik. Die frühe Diagnose der genetischen Veranlagung des Patienten ermöglicht auf der einen Seite prophylaktische Maßnahmen und eine frühe unterstützende Therapie. Auf der anderen Seite werden Angehörige und Patient möglicherweise schon im Kindesalter des Betroffenen mit der Wahrscheinlichkeit des Ausbruchs einer lebensbedrohlichen Krankheit in mehreren Jahrzehnten konfrontiert. Risiken und Nutzen müssen vor einer Diagnose daher sorgfältig abgewägt werden.[21]
Bei Patienten mit familiärer Veranlagung (Prädisposition) kann die Diagnose per Sonographie ab dem 20. Lebensjahr gestellt werden, wenn pro Niere mindestens zwei Nierenzysten nachweisbar sind. Fehlende Zysten schließen dagegen bei über 30-jährigen die Erkrankung aus.[22][23]
Pathogenese
Die Entstehung und Entwicklung von Zystennieren, die Pathogenese, beruht auf einer zystischen Degeneration der sogenannten Tubuli (Harnkanälchen) in den Nieren. Diese führt bei der autsomal-dominant-vererbten PKD im Verlauf von Jahrzehnten zu einer zunehmenden Vergrößerung der Nieren. Es kann dabei zu einer Funktionseinschränkung bis hin zum völligen Verlust der Nierenfunktion kommen. Beide Nieren sind gleichmäßig betroffen. Mehrere hundert Zysten, die in ihrer Erscheinung prall elastisch sind, können dabei pro Organ ausgebildet werden. Masse und Volumen der Nieren können dadurch erheblich anwachsen. Während eine gesunde Niere durchschnittlich eine Masse von 160 g aufweist, können polyzystische Nieren bis zu 8 kg bei bis zu 40×25×20 cm3 (= 20 Liter) Volumen erreichen (gesunde Niere: 12×6×3 cm3 = 0,216 Liter). Trotz des erheblich gesteigerten Platzbedarfs des Organs kommt es nur relativ selten zu Funktionsstörungen der benachbarten Organe.[24]
Die Zysten finden sich sowohl am Nierenmark (Medulla renis), als auch an der Nierenrinde (Cortex renalis). Prinzipiell kann dabei jeder Bereich eines Nephrons eine Zyste ausbilden. Bevorzugt betroffen sind jedoch die Glomeruli und die Henlesche Schleife. Gefüllt sind die Zysten mit dem sogenannten Tubulusharn. Der Durchmesser einer einzelnen Zyste kann von wenigen Millimetern bis über 100 mm sehr stark variieren. Große Zysten können so mehrere hundert Milliliter Tubulusharn enthalten. Das Innere der Zysten besteht aus einem einschichtigen Plattenepithel oder einschichtigem isoprismatischen (kubischen) Epithel. Mit dem Fortschreiten der Erkrankung können sowohl die Anzahl, als auch die Größe der vorhandenen Zysten zunehmen.[2]
Ätiologie
Polyzystische Veränderungen in den Nieren sind ein Krankheitsbild, das bei einer Reihe von Erkrankungen auftritt. Sie können als Abweichung von der normalen Entwicklung der Nieren sporadisch entstehen oder im erwachsenen Leben erworben werden („erworbene Zystennieren“). Die weitaus häufigere Ursache (Ätiologie) für diese Erkrankung sind aber durch Vererbung übertragene Defekte in bestimmten Genen („hereditäre Zystennieren“). Den mit Abstand größten Anteil hat dabei die autosomal-dominante polyzystische Nierenerkrankung (engl.: autosomal-dominant polycystic kidney disease, ADPKD).[25] Diese Erkrankung ist die häufigste erbliche Ursache eines chronischen Nierenversagens: Etwa 7 % aller Dialysepatienten leiden an ihr.[26]
Daneben verursachen verschiedene andere – erheblich seltenere – Erbkrankheiten Polyzystennieren. Auch erworbene Zystennieren können sich – vor allem bei Dialysepatienten – einstellen.[27] Da der weitaus größte Teil von Zystennieren durch die ADPKD hervorgerufen wird, findet der Begriff „Zystenniere“ oft eine synonyme Anwendung für die ADPKD.
Erbliche Zystennieren
Die Mehrzahl polyzystischer Nierenerkrankungen ist erblich (hereditär) bedingt. Dabei kann eine Vielzahl von verschiedenen Genen betroffen sein und so die Krankheit auslösen. Die nachfolgend aufgeführten Syndrome stellen eine Auswahl der wichtigsten erblich bedingten polyzystischen Nierenerkrankungen dar.[28] Ein Teil der Erkrankungen wird dem sogenannten NPH-MCKD-Komplex zugerechnet.
Übersicht über hereditäre polyzystische Nierenerkrankungen[22]
Gen Chromosom
GenlocusProtein Erkrankung Inzidenz Alter *) Autosomal-dominant PKD1 16 p13.3 Polycystin-1 ADPKD 1:500-1000 ca.50 PKD2 4 q21-q23 Polycystin-2 ADPKD 1:3500-7000 ca.70 VHL 3 p26-p25 VHL30 Von-Hippel-Lindau 1:35.000 20-30 TSC1 9 q34 Hamartin Tuberöse Sklerose 1:10.000 (beide zusammen) 30-40 TSC2 16 p13.3 Tuberin Tuberöse Sklerose ? 1 q21 Medullär-zystische Nierenerkrankung Typ 1 1 bis 9 : 1.000.000 (Typ 1+2)[29] 62 UMOD 16 p12.3 Uromodulin Medullär-zystische Nierenerkrankung Typ 2 32 Autosomal-rezessiv PKHD1 6 p21.2-p12 Fibrocystin ARPKD 1:20.000 <20 NPHP1 2 q13 Nephrocystin-1 Nephronophthise (juvenile) ca. 1:100.000 (alle NPHP) 13 NPHP2 9 q22-q31 Inversin Nephronophthise (infantile) <1 NPHP3 3 q22.1 Nephrocystin-3 Nephronophthise (adoleszente) 19 NPHP4 1 p36.22 Nephroretinin Nephronophthise 21 NPHP5 Nephrocystin 5 Nephronophthise 13 NPHP6 Nephrocystin 6 Nephronophthise GLIS2 16 p13.3 GLI-Similar Protein 2[30] Nephronophthise BBS1 11 q13.1 BBS1-Protein Bardet-Biedl-Syndrom 1:140.000 (alle BBS) BBS2 16 q21 BBS2-Protein Bardet-Biedl-Syndrom ARL6 3 p13-p12 BBS3-Protein
ADP-ribosylation factor-like protein 6Bardet-Biedl-Syndrom BBS4 15 q22.3-q23 BBS4-Protein Bardet-Biedl-Syndrom BBS5 2 q31.1 BBS5-Protein Bardet-Biedl-Syndrom MKKS 20 p12 BBS6-Protein McKusick-Kaufman-Syndrom BBS7 4 q27 BBS7-Protein Bardet-Biedl-Syndrom TTC8 14 q31.3 BBS8-Protein Tetratricopeptide Repeat Domain 8 BBS9 7 p14 PTHB1 Bardet-Biedl-Syndrom BBS10 12 q21.2 BBS10-Protein Bardet-Biedl-Syndrom TRIM32 9 q33.1 Zinkfingerprotein HT2A Tripartite motif-containing 32 BBS12 4 q27 BBS12-Protein Bardet-Biedl-Syndrom X-chromosomal-dominant CXORF5 X p22.3-p22.2 OFD1 Oro-fazio-digitales Syndrom Typ 1 1:250.000 Unbekannter Erbgang ? ? Markzystenniere 1:5000 40-50 ? ? Multizystische Nierendysplasie <10 und 50-60 *) mittleres Alter bis zur terminalen Niereninsuffizienz
Autosomal-dominante polyzystische Nierenerkrankung
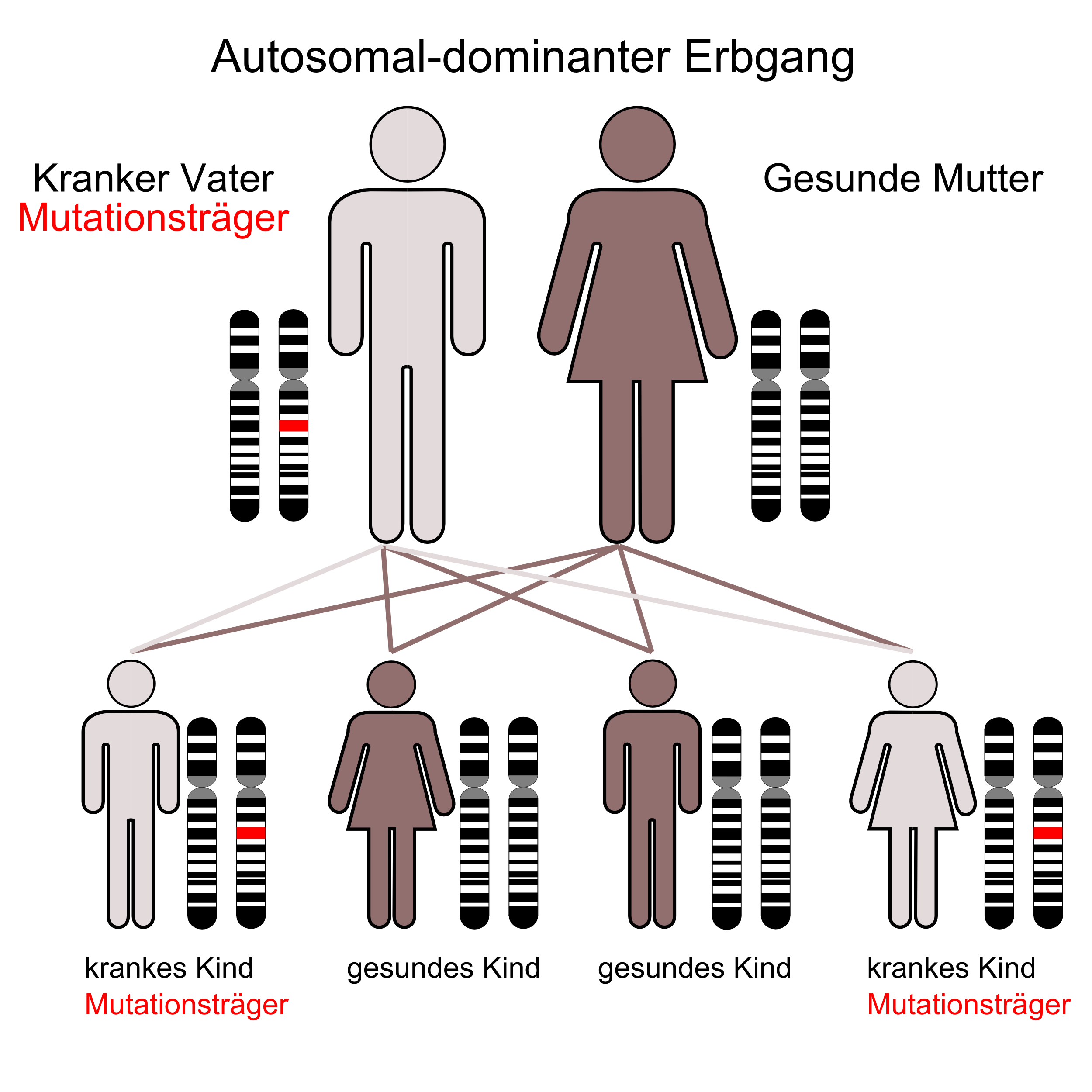
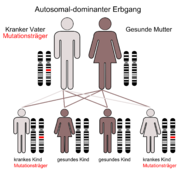 Der autosomal-dominante Erbgang
Der autosomal-dominante ErbgangDie autosomal-dominante polyzystische Nierenerkrankung (ADPKD), auch zystische Nierendegeneration Potter Typ III[31] genannt, ist die häufigste lebensbedrohende Erbkrankheit beim Menschen. Weltweit gibt es etwa 5 Millionen von der ADPKD betroffene Menschen. Die Inzidenz liegt bei 1:500 bis 1:1000.[25] In den USA ist die Erkrankung beispielsweise zweimal häufiger als Multiple Sklerose und zehnmal häufiger als Sichelzellenanämie.[2] Männer und Frauen sind gleich häufig betroffen. Ebenso spielen Rasse und Herkunft keine Rolle. Die Symptome werden in der Regel erst im Erwachsenenalter beobachtet. Der Erbgang der ADPKD ist autosomal-dominant (monogenetisch)[32] mit vollständiger Penetranz.[33] [34] Bedingt durch den autosomal-dominanten Erbgang erbt im statistischen Mittel die Hälfte der Kinder von ihren Eltern das mutierte Gen und wird selbst an ADPKD erkranken. Etwa 50 % aller Mutationsträger erleiden eine progressive Niereninsuffizienz.[22] Im Alter von durchschnittlich 58 Jahren ist bei der Hälfte der ADPKD-Patienten eine Nierenersatztherapie indiziert.[35]
Als systemische Erkrankung sind bei der ADPKD häufig auch andere Organe – in den meisten Fällen die Leber – von einer Zystenbildung betroffen.[36] Je nach Autor haben bis zu 75 % der von der ADPKD Betroffenen Leberzysten.[2]
Genetik
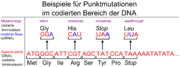 Beispiele für Punktmutationen
Beispiele für PunktmutationenBisher wurden Mutationen in zwei verschiedenen Genen als Ursache der Erkrankung bei ADPKD-Patienten nachgewiesen: Die Gene PKD1 und PKD2. PKD1 liegt beim Menschen auf Chromosom Genlocus p13.3-p13.12. Es kodiert das Protein Polycystin-1. Bei Patienten mit signifikanten Mutationen in PKD1 erreicht die Niereninsuffizienz im Alter von durchschnittlich 50 Jahren ihr Endstadium, womit eine Nierenersatztherapie angezeigt ist. Patienten mit Mutationen in PKD2, das auf Chromosom 4 Genlocus q21-q23 liegt und Polycystin-2 kodiert, erreichen dieses Stadium erheblich später, im Alter von durchschnittlich 70 Jahren (late onset). Etwa 85 % der Patienten mit ADPKD tragen eine oder mehrere Mutationen in PKD1. Die restlichen circa 15 % entfallen auf Mutationen in PKD2.[37]
Auf zellulärer Ebene betrachtet handelt es sich bei der ADPKD um einen rezessiven Mechanismus. Eine Bedingung für die Krankheit ist als erstes eine Keimbahnmutation in einem der PKD1- beziehungsweise PKD2-Allele. Als zweites muss eine somatische Mutation, ein sogenannter second hit stattfinden, damit die Zystenbildung initiiert wird.[38][39] Dieser Verlust der Heterozygotie (loss of heterocygosity, LOH) findet bei der ADPKD offensichtlich immer statt.[40] Die initiale somatische Mutation kann auf dem jeweils anderen Gen liegen. In diesem Transheterozygotie genannten Fall ist die Keimbahnmutation auf PKD1 und die somatische Mutation auf PKD2, beziehungsweise umgekehrt.[41] Im Tiermodell wurde festgestellt, dass Keimbahnmutationen die beiden Allele eines PKD–Gens betreffen perinatal tödlich sind.[42] Mit dem second hit verliert die betroffene Zelle die Fähigkeit die Proliferation zu hemmen und wird so der Ausgangspunkt für die Bildung einer neuen Zyste.[22] Ein wichtiges Indiz für die Richtigkeit der Second-hit-Theorie sind Versuche mit Knockout-Mäusen, bei denen PKD1 beziehungsweise PKD2 abgeschaltet (Gendeletion) wurden. So erkranken nur homozygote Tiere, während die Heterozygoten eine nahezu normale Entwicklung nehmen.[43][44] Die Second-hit-Theorie dient auch als Erklärung dafür, dass nur etwa 1 % aller Nephrone bei der ADPKD Zysten bilden, obwohl alle Zellen die vererbte Mutation tragen.[22]
Ab 1995 vermutete man noch ein drittes Gen, PKD3 genannt, als weitere mögliche Ursache für die ADPKD.[45][46] Später wurden bei vier weiteren Familien mit Zystennieren aus verschiedenen Ländern Mutationen beobachtet, die ihre Ursache weder in PKD1 noch in PKD2 hatten.[47][48] Die Existenz dieses Gens wird mittlerweile bezweifelt.[49][50][51][52]
Die Häufigkeit der verschiedenen Mutationstypen bei der ADPKD:[53]
Gen Loci Exons Mutationstyp Häufigkeit (%) PKD1 16p13.3 46 (14.1 kb) Nonsense 33 Frameshift 28 In-frame 6 Splicing 14 Missense 19 PKD2 4q21–q23 15 (5 kb) Nonsense 37 Frameshift 39 Splicing 17 Missense 6 Deletion 1 Molekulare Ursachen und Zystenbildung
 Schematische Darstellung von PKD1 und PKD2 an einer Zelle.[54]
Schematische Darstellung von PKD1 und PKD2 an einer Zelle.[54]Die von den betroffenen Genen codierten Proteine Polycystin-1 und Polycystin-2, sowie das vom PKHD1-Gen kodierte Fibrocystin, liegen an der Basis des primären Zilium der Zellen des Nierenkanälchen (Nierentubuluszellen). Das Primärzilium ist ein haarfeiner Zellfortsatz, von dem jede Zelle jeweils nur einen einzigen ausbildet. Nach den gegenwärtigen Erkenntnissen spielt bei allen zu Zystennieren führenden Erkrankungen eine Fehlfunktion des Primärziliums die entscheidende Rolle für die Ausbildung von Zysten.[53][55] Die Primärzilien der Tubuluszellen ragen in das Tubuluslumen und dienen dort wahrscheinlich der Wahrnehmung der Flüssigkeitsströmung. Zudem ist das Primärzilium bei der Zellteilung an der räumlichen Ausrichtung der Mitosespindel beteiligt.[56] Die beiden Polycystine bilden einen Calcium-regulierenden Ionenkanal, der für Calcium-Ionen durchlässig ist. Der Polycystin-Komplex spielt mit mehreren Signalwege und mechano-sensorischen Funktionen eine wichtige Rolle im Primärzilium. Die physiologische Funktion dieses Zellorganells ist bisher noch weitgehend unverstanden.[57]
Der Ursprung der Zysten kann in jedem Abschnitt eines Nephrons – vom Glomerulum bis zu den Sammelgängen (Tubulus renalis colligens) – seinen Ausgangspunkt haben. Erreichen die Zysten einen Durchmesser von über 0,2 mm, so haben sie keine Verbindung mehr zu den Nierenkanälchen (Tubuli).[58][20]
Damit sich die Zysten ausbilden können, muss sich die Anzahl der Zellen innerhalb der Zystenwand erhöhen. Dies geschieht durch eine exzessive Proliferation der Epithelzellen der Nieren. Dabei ist das Protein mTor (mammalian Target of Rapamycin) hochreguliert. Im Zystenlumen muss sich außerdem, durch eine erhöhte Sekretion und/oder einen verminderten Abfluss, Flüssigkeit ansammeln. Diese transepitheliale Flüssigkeitssekretion ist abhängig von der sekundär aktiven Chloridionen-Sekretion. Die Chloridionen-Sekretion wird über den CFTR (cystic fibrosis transmembrane conductance regulator) oder über einen kalziumabhängigen Chloridkanal geregelt. Beide befinden sich in der apikalen Zellmembran.[20]
Verlauf und Prognose
Der Verlauf der ADPKD ist langsam progredient (fortschreitend). Bereits vor dem Einsetzen der Niereninsuffizienz ist bei den betroffenen Patienten eine Störung der Harnkonzentrierung (Wasserrückresorption) feststellbar.[11][59][31] Die Nierenfunktion erfährt im Anfangsstadium der Erkrankung durch die Zystenbildung keine Einschränkung. Erst ab einer Nierengröße von 1000 cm3 nimmt die Leistung ab. Liegt das Nierenvolumen oberhalb von 1500 cm3, so reduziert sich die glomeruläre Filtrationsrate jährlich um etwa 4 bis 5 ml·min-1. Durchschnittlich nimmt das Volumen der Nieren bei Patienten mit einem Nierenvolumen über 750 cm3 pro Jahr um über 5 % zu.[22] Die ersten Symptome der Erkrankung werden meist im Alter zwischen 30 und 40 Jahren wahrgenommen. Allgemein liegt hier jedoch eine große Variationsbreite – oft auch innerhalb einer Familie – vor.[60]
In fast allen Fällen führt die Erkrankung zur terminalen Niereninsuffizienz (endgültiges Nierenversagen). Frauen erreichen dieses Stadium durchschnittlich sechs Jahre später als Männer.[61]
Ein weiteres Überleben ist dann nur noch durch eine Nierenersatztherapie, das heißt Dialyse oder Nierentransplantation, gewährleistet. Es ist noch nicht vollständig geklärt, warum polyzystische Nieren letztlich zur terminalen Niereninsuffizienz führen. Über die Druckatrophie des Parenchyms alleine lässt sich der Mechanismus nicht erklären. Chirurgische Eingriffe wie beispielsweise Punktionen bewirken keine Verzögerung des Krankheitsverlaufes. Aus histologischen Untersuchungen lässt sich schließen, dass die Hypertonie ein wichtiger Faktor für die Progression der Niereninsuffizienz ist.[2]
Neben der Genetik hat auch die Umgebung und die Lebensweise des Patienten einen Einfluss auf den Verlauf der ADPKD. Bei Frauen wurde beispielsweise festgestellt, dass mehrere Entbindungen, sowie andere estrogene Faktoren den Krankheitsverlauf erheblich verschlechtern.[62] Das im Vergleich zu Frauen beschleunigte Wachstum der Zysten und das frühzeitigere Erreichen des terminalen Nierenversagens bei Männern, wird ebenfalls auf hormonelle Einflüsse zurückgeführt.[63][64] Auch das Tabakrauchen beeinflusst – insbesondere bei Männern – die Progression der ADPKD negativ. Eine mögliche Erklärung sind hierbei die bekannten negativen Effekte des Rauchens auf die Blutgefäße. [65][66][52]
Lebenserwartung
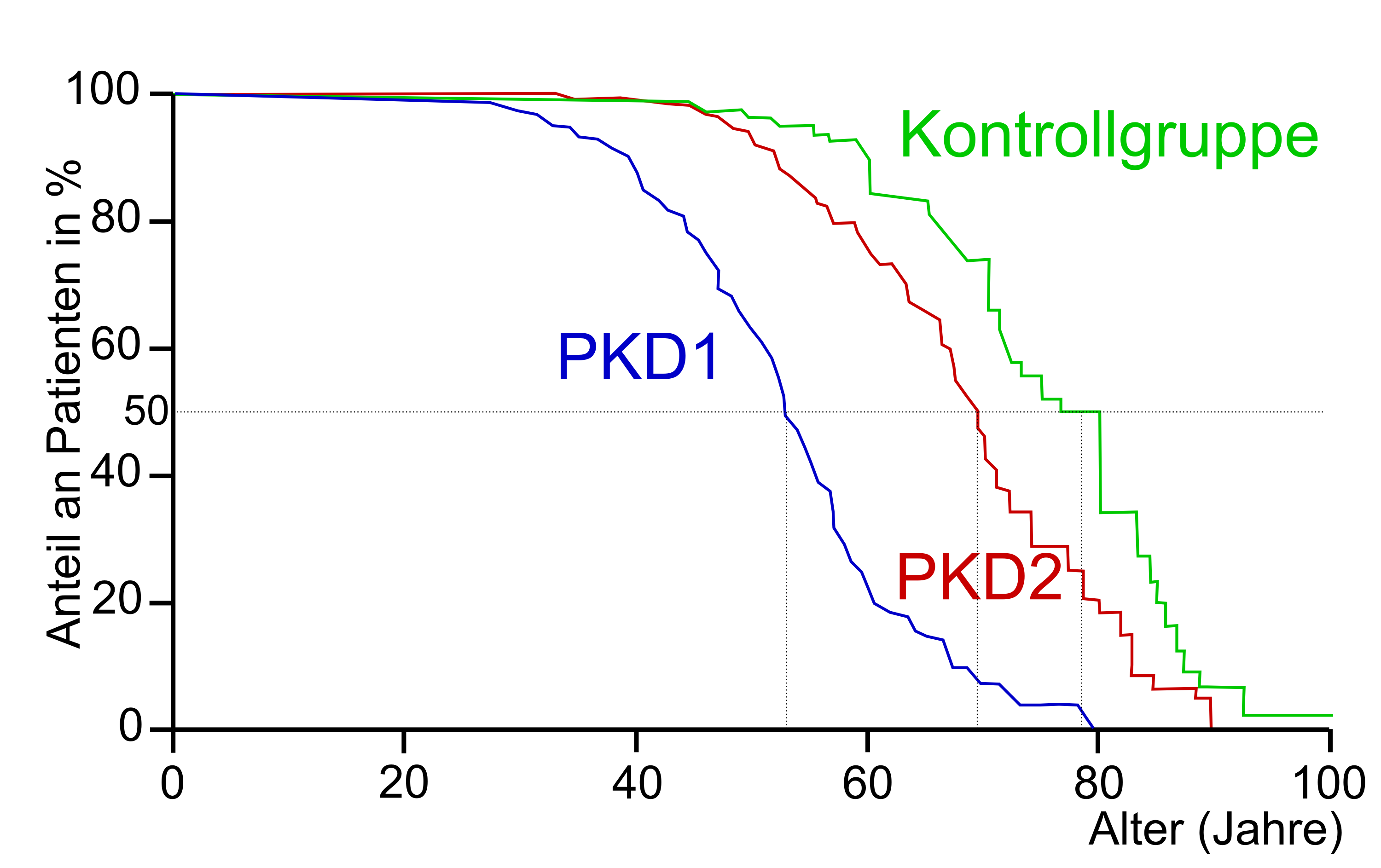
 Vergleich der Überlebensrate von Patienten mit ADPKD. In Blau: Mutationen in PKD1. In Rot: Mutationen in PKD2. In Grün: Kontrollgruppe ohne ADPKD.[67]
Vergleich der Überlebensrate von Patienten mit ADPKD. In Blau: Mutationen in PKD1. In Rot: Mutationen in PKD2. In Grün: Kontrollgruppe ohne ADPKD.[67]In einer Studie wurden 333 Patienten aus 31 Familien mit PKD1 und 291 Patienten mit PKD2 aus ebenfalls 31 Familien, mit einer 398 Personen starken, geografisch identischen, Kontrollgruppe verglichen. PKD1-Patienten erreichten ein mittleres Alter von 53,0 Jahren (±1,8 Jahre; 95 % Wahrscheinlichkeit). PKD2-Patienten kamen dagegen auf durchschnittlich 69,1 Jahre (±2,2 Jahre; 95 %), während die Personen aus der Kontrollgruppe 78,0 Jahre (±4,2 Jahre; 95 %) wurden (siehe dazu nebenstehende Grafik).[67]
Todesursachen
In einer retrospektiven Studie wurde die Todesursache von 129 Patienten mit ADPKD analysiert. Danach verstarben 36 % an einer Herzerkrankung und 24 % an Infektionen. Bei den Infektionen lag in 94 % der Fälle eine Sepsis (Blutvergiftung) vor. Bei den Obduktionen wurde bei 89 % aller Patienten eine Herzhypertrophie und bei 81 % eine Koronare Herzkrankheit festgestellt. Ein neurologisches Ereignis führte bei 12 % der Patienten und die Ruptur eines Hirn-Aneurysma bei 6 % zum Tod. Durch Bluthochdruck bedingte Hirnblutungen waren in 5 % und ein ischämischer Schlaganfall bei 1 % der Patienten die Todesursache. Kein Patient verstarb an Nierenkrebs.[68]
Autosomal-rezessive polyzystische Nierenerkrankung
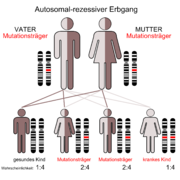 Der autosomal-rezessive Erbgang
Der autosomal-rezessive ErbgangDie autosomal-rezessive polyzystische Nierenerkrankung (ARPKD), auch als Schwammniere oder Potter I-Niere bezeichnet, manifestiert sich bereits in der Kindheit. Die Prävalenz dieser Erkrankung liegt bei Neugeborenen im Bereich von 1:6000 bis 1:40.000, durchschnittlich bei 1:20.000. Die Erkrankung ist somit relativ selten. Die Penetranz ist vollständig.[69] Etwa jeder siebzigste Mensch ist Träger der Mutation (siehe Grafik autosomal-rezessiver Erbgang).[70] Mutationen im PKHD1–Gen – das beim Menschen auf Chromosom 6, Genlocus p21.1-p12 liegt – können zur Ausbildung von Zystennieren führen.[71][72][73] Das von PKHD1 kodierte Protein Fibrocystin findet sich zusammen mit Polycystin-2 im Basalkörper der primären Zilien. In der apikalen Domäne polarisierter epithelialer Zellen ist es offensichtlich in die Bildung der Tubuli und/oder der Aufrechterhaltung der Architektur des Lumens des Sammelrohrs involviert.[74] Dem entsprechend sind bei der ARPKD im wesentlichen die Sammelrohre von der Zystenbildung betroffen.[2]
Die ARPKD manifestiert sich bei Patienten bereits in sehr jungen Jahren (early onset). Der Altersbereich beträgt 0 bis 20 Jahre. Die mittlere Lebenserwartung der betroffenen Kinder beträgt sechs Jahre. Man unterscheidet zwischen perinataler (28. Schwangerschaftswoche bis sieben Tage nach der Geburt), neonataler (neugeboren), infantiler (kindlich) und juveniler (jugendlich) Form. Je geringer das Alter der Manifestation ist, um so schlechter ist dabei die Prognose.[2]
NPH-MCKD-Komplex
- → siehe Hauptartikel NPH-MCKD-Komplex, Medullär-zystische Nierenerkrankung Typ 2, Medullär-zystische Nierenerkrankung Typ 1, Nephronophthisis
Der NPH-MCKD-Komplex (nephronophthisis-medullary cystic kidney disease) ist eine Gruppe von genetisch bedingten Erkrankungen der Niere, die zu einer Zystenniere führt. Der Erbgang ist im Fall der Nephronophthisis autosomal-rezessiv, während er bei den beiden Formen der medullär-zystischen Nierenerkrankung autosomal-dominant ist. Die Erkrankungen haben als gemeinsames Krankheitsbild die Ausbildung von Zystennieren an der Rinde-Mark-Grenze (kortikomedulläre Grenze). Alle Krankheiten des NPH-MCKD-Komplexes führen in Abhängigkeit vom betroffenen Gen in bestimmten Altersbereichen zum terminalen Nierenversagen.
Bardet-Biedl-Syndrom
- → siehe Hauptartikel Laurence-Moon-Biedl-Bardet-Syndrom
Das Bardet-Biedl-Syndrom (BBS) ist eine sehr seltene oligogenetische[75] Erbkrankheit mit autosomal-dominantem Erbgang. Die Ursache der Erkrankung sind Mutationen auf den BBS-Genen 1 bis 12. Neben der Ausbildung von polyzystischen Nieren[22][76] kommt es zu einer Degeneration der Netzhaut, kindlicher Adipositas, geistiger Behinderung, Missbildungen des Harn- und Geschlechtsapparates und Polydaktylie (Vielfingerigkeit).[77]
Tuberöse Sklerose
- → siehe Hauptartikel Tuberöse Sklerose
Bei der autosomal-dominant vererbten tuberösen Sklerose sind einzelne Nierenzysten häufig. Seltener tritt auch eine polyzystische Nierenerkrankung auf. Ursache hierfür sind meist größere Deletionen, die sowohl das bei der tuberösen Sklerose betroffene TSC2-Gen als auch das PKD1-Gen betreffen; beide Gene sind in enger Nachbarschaft auf Chromosom 16 lokalisiert.[78]
Oro-fazio-digitales Syndrom Typ 1 (OFD 1)
 Der X-chromosomal-dominante Erbgang
Der X-chromosomal-dominante Erbgang- → siehe Hauptartikel Oro-fazio-digitales Syndrom Typ 1
Das Oro-fazio-digital Syndrom Typ 1, auch Papillon-Leage-Psaume-Syndrom genannt, ist eine sehr seltene X-chromosomal-dominant vererbte Krankheit. Die Prävalenz liegt bei Neugeborenen bei etwa 1:250.000. Die Krankheit weist eine Reihe unterschiedlicher Symptome, vor allem im Gesichts- und Mundbereich auf, sowie der bei vielen Patienten zu beobachtenden Neigung zu polyzystischen Nieren.[79] Letztere werden meist oft sehr spät diagnostiziert, wenn die Niereninsuffizienz schon weit fortgeschritten ist.[80]
Die Erkrankung ist für das männliche Geschlecht pränatal tödlich.[81]
Beim Oro-fazio-digitales Syndrom Typ 2, OFD2 beziehungsweise Mohr-Syndrom genannt, werden keine Veränderungen an den Nieren beobachtet.
Erworbene Zystennieren
Eine besondere Form der Endstadiumniere, die als sekundäre polyzystische Transformation oder auch als erworbene Zystenniere (engl.: acquired cystic kidney disease, ACKD) bezeichnet wird, entwickelt sich bei 40 bis 50 % aller Patienten nach Langzeitdialyse.[82] Sie ist eine sehr ernst zu nehmende Komplikation beim terminalen Nierenversagen (ESRD).[83] Bei Transplantierten können dabei sowohl die eigenen Nieren als auch das Transplantat betroffen sein.[84] Ursache für die Ausbildung der erworbenen dialysebedingten Zystennieren ist meist eine mehrjährige Dialyse wegen Analgetikanephropathie. Bei Patienten mit terminaler Niereninsuffizienz treten Zysten an den Nieren sehr häufig auf. Häufigkeit und Zystengröße wachsen mit zunehmender Dialysedauer an.[85] Von der Erkrankung sind beide Geschlechter gleich häufig betroffen, wobei das Alter der Patienten keine Rolle spielt. Die Wahrscheinlichkeit, dass sich als weitere Komplikation eine Nierenkrebserkrankung einstellt, ist signifikant – insbesondere bei männlichen Patienten – erhöht.[83]
Therapie
 Sirolimus (Rapamycin)
Sirolimus (Rapamycin)Derzeit gibt es kein zugelassenes Medikament zur Behandlung einer polyzystischen Nierenerkrankung. Bei etwa 50 % aller ADPKD-Patienten – die das Gros der Patienten mit einer polyzystischen Nierenerkrankung bilden – wird im Laufe ihres Lebens eine Nierenersatztherapie notwendig. Eine Heilung ist nur durch eine Nierentransplantation möglich.
Adjuvante Maßnahmen
Der Einstellung des arteriellen Blutdruckes, meist mit Hilfe von ACE-Hemmern, hat als adjuvante Maßnahme eine besondere Bedeutung bei polyzystischen Nieren. Darüber hinaus gibt eine Reihe von Empfehlungen für Patienten mit Zystennieren, die zwar ebenfalls keine Heilung ermöglichen, aber die Krankheitsfortschritt (Progression) für den Patienten günstiger gestalten.
Da Koffein im Verdacht steht das Zystenwachstum zu beschleunigen,[86] sollten Patienten auf koffeinhaltige Getränke möglichst verzichten.[2] Eine salzarme Diät hilft den Blutdruck zu senken, der mit einer gestörten Ausscheidung von Natrium-Ionen in Zusammenhang steht.[87] Nichtsteroidale Antirheumatika, Mischanalgetika, bestimmte Antibiotika und andere nierentoxische Medikamente sollten weitgehend gemieden werden. Zysteninfektionen werden dagegen möglichst frühzeitig mit zysten- beziehungsweise gallengängigen Antibiotika behandelt.[88]
Nierenersatztherapie
 Ein Hämodialysator
Ein Hämodialysator- → siehe Hauptartikel Nierenersatztherapie
Nur die Nierenersatztherapie, das heißt Dialyse oder Nierentransplantation, sichert nach der terminalen Niereninsuffizienz das Überleben des Patienten. In den meisten Fällen erfolgt die Dialyse in Form der Hämodialyse, da durch die übergroßen Nieren – und oft auch Leber – der Bauchraum sehr beengt ist und eine Peritonealdialyse somit nicht möglich ist. Die Nierentransplantation ist – wenn möglich – der Dialyse vorzuziehen. Sie ermöglicht die Wiederherstellung der körperlichen Leistungsfähigkeit, der Lebensqualität und der sozialen Integration der Patienten. Sie verbessert außerdem die Lebenserwartung gegenüber der Dialyse erheblich. Problematisch sind die langen Wartezeiten auf Spendernieren, bedingt durch die geringe Anzahl an verfügbaren Spendernieren.
Polyzystische Nieren werden – gegenüber der früheren üblichen Praxis – nur in Ausnahmefällen, wenn beispielsweise die Nierenvolumina eine kritische Größe angenommen haben, prätransplantativ entfernt.[89][2][90]
Zukünftige Therapieansätze
Die Behandlung von Patienten mit Zystennieren verursacht alleine in den USA jährliche Kosten in Höhe von über 1 Mrd US$.[91] Diese Summe ergibt sich im wesentlichen aus den Kosten für die nach dem terminalen Nierenversagen notwendige Nierenersatztherapie.
Vermehrung und Größenzunahme der dünnwandigen, flüssigeitsgefüllten Zysten hängt zwei Prozessen ab: Proliferation von Zellen des Zystenepithels und Sekretion von Flüssigkeit in die Zysten. Beide Prozesse sind von cAMP abhängig. cAMP stimuliert den Ras/MAP-Kinase-Weg und führt so zu einem abnormalen Zellwachstum. Zudem aktiviert cAMP den CFTR-Chloridkanal und fördert so die Flüssigkeitssekretion in die Zysten. [92] Derzeit in Erprobung befindliche Therapieansätze setzen an beiden cAMP-abhängigen Prozessen an, um Zystenbildung und -wachstum zu verlangsamen.
Bildgebende Verfahren und Untersuchung neuer Therapieansätze
Das Durchschnittsalter bei Diagnosestellung der ADPKD liegt derzeit bei 27 Jahren. Wenn eine Nierenfunktionseinschränkung eintritt, kommt es zu einer raschen Abnahme der GFR von ~5.9 ml/min pro Jahr. Bislang war keine randomisierte Studie in diesem späten Stadium der Erkrankung in der Lage, den günstigen Effekt einer Behandlung nachzuweisen. Wegen der langen präsymptomatischen Phase und des späten Auftretens der Niereninsuffizienz sind die primären Endpunkte, welche üblicherweise bei Studien zu chronischen Nierenerkrankungen untersucht werden, wie Zeit bis zur Dialysebehandlung, Verdoppelung des Serum-Kreatinins oder Tod, bei Studien zu polyzystischen Nierenerkrankungen nur bedingt brauchbar.[93] Aus diesem Grund wurde das Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) gegründet, dessen Aufgabe es ist, bildgebende Verfahren zu untersuchen, die in den Frühstadien Aussagen zum Erkrankungsverlauf ermöglichen. Ein wichtiges Ergebnis der Untersuchungen des CRISP-Consortiums mittel Magnetresonanztomographie ist, dass bei Patienten mit ACPKD die Zysten kontinuierlich und quantifizierbar wachsen und das Zystenwachstum mit der Abnahme der Nierenfunktion korreliert: Eine stärkere Zunahme der Zystengröße ist mit einer schnelleren Abnahme der Nierenfunktion assoziiert.[94]
HALT-Polycystic Kidney Disease (HALT-PKD) ist eine prospektive Studie, mit der aktuell die Auswirkungen einer Blockade des Renin-Angiotensin-Aldosteron-Systems und/oder einer strikten Blutdruck-Kontrolle im Frühstadium der Erkrankung auf das Zystenwachstum untersucht wird, bzw. in späteren Stadien der Erkrankung die Auswirkung auf Verdopplung des Serum-Kreatinins, Dialysebeginn und Tod.
Hemmung der Zellproliferation
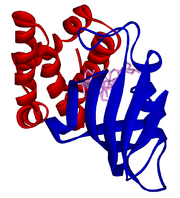 Modell der Rapamycin-bindenden Domäne von mTOR (rot) in einem Komplex mit Rapamycin (Pink) and FKBP12 (blau).
Modell der Rapamycin-bindenden Domäne von mTOR (rot) in einem Komplex mit Rapamycin (Pink) and FKBP12 (blau).In den letzten Jahren konnten mit dem zunehmenden molekularbiologischen Wissen über die Ursachen der PKD neue Therapieansätze gefunden werden. Einige dieser Ansätze befinden sich zur Zeit in der klinischen Erprobung. Initiierend war dabei aber eher ein Zufallsbefund: bei einigen Patienten, die eine Fremdniere erhalten hatten, wurde in einer retrospektiven Studie festgestellt, dass die verbliebene polyzystische Niere an Volumen nicht weiter zunahm, sondern teilweise sich die Zysten etwas zurückbildeten.[95] Die Anzahl der Patienten in der ersten Studie war mit n=4 statistisch zwar nicht aussagekräftig, in verschiedenen Tiermodellen konnte der Effekt jedoch statistisch sicher nachgewiesen werden.[96][97] Die offensichtliche Ursache für diese Verbesserung war die Einnahme von Sirolimus (Rapamycin), das den Patienten als Immunsuppressivum verabreicht wurde. Patienten mit einer Spenderniere müssen, um eine Abstoßung des Fremdorgans durch das körpereigene Immunsystem zu vermeiden, zeitlebens Immunsuppressiva einnehmen.[98] Derzeit laufen in verschiedenen Ländern breit angelegte klinische Studien zur Überprüfung der Wirksamkeit von Sirolimus für eine mögliche Therapie polyzystischer Nieren.[99][100]
Neben Sirolimus und Derivaten dieser Verbindung, wird auch an anderen potenziellen Substanzen geforscht, die zum Teil andere Signalwege nutzen. So sind beispielsweise die cAMP-Antagonisten Somatostatin[101] und Vasopressin[101] potenzielle Wirkstoffe, da erhöhte Werte von cAMP die Proliferation und Sekretion zystischer Epithelzellen stimulieren.[102][103]
Triptolide ist ein kleines Molekül, das aus einem traditionellen chinesischen Medikament (Thunder God Vine) isoliert wurde, und das anti-proliferative und pro-apoptotische Eigenschaften aufweist. Triptolide fördert die Calcium-Freisetzung durch einen Polycystin-2 abhängigen Stoffwechselweg und hemmt im Tiermodell Zystenbildung und Zystenwachstum.[104][105][106][107]
Hemmung der Flüssigkeitssekretion
Bei Patienten mit polyzstischer Nierenerkrankung sind die Spiegel von antidiuretischem Hormon (Vasopressin) erhöht. Der V2-Vasopressin-Rezeptor wird in distalem Tubulus und Sammelrohr exprimiert, der Stelle des Nephron, an der die Zystenbildung stattfindet. Vasopressin stimuliert über den V2-Rezeptor die Bildung von cAMP im distalen Tubulus.[108]
Im Tiermodell hemmen V2-Rezeptor-Antagonisten die Bildung von cAMP, Größenzunahme der Nieren und Zystenbildung und schützen die Nierenfunktion. [109]
Der V2-Rezeptor-Antagonist Tolvaptan erwies sich bei Patienten mit ADPKD in Phase 2-3 Studien als sicher und gut verträglich. Eine Placebo-kontrollierte Doppelblindstudie an Patienten mit ADPKD, normaler Nierenfunktion und einem Nierenvolumen über 750 ml wird derzeit durchgeführt.
Komplikationen
 Nierenstein in eine computertomografischen Aufnahme (siehe roter Pfeil)
Nierenstein in eine computertomografischen Aufnahme (siehe roter Pfeil)Typische Komplikationen bei Zystennieren sind Blutdruckerhöhung durch Stimulation des Renin-Angiotensin-Aldosteron-Systems und Harnwegsinfekte.
Von den Harnwegsinfekten sind – bedingt durch die kürzeren Harnwege – insbesondere weibliche Patientinnen stark betroffen. In den meisten Fällen handelt es sich um Infektionen der Harnblase durch gramnegative und nosokomiale Keime. Die Behandlung der Harnwegsinfekte erfolgt symptomatisch, vorzugsweise mit lipophilen Antibiotika. Extreme Infektionen, wie beispielsweise eine Pyonephrose (eine eitrige Hydronephrose), können zur Entfernung der betroffenen Niere (Nephrektomie) führen.[2]
Während die Häufigkeit von Nierensteinen in der Bevölkerung bei etwa 5 % liegt, sind 10 bis 34 % der Patienten mit polyzystischen Nieren von diesen Ablagerungen betroffen. Eine mögliche Ursache für das erhöhte Vorkommen von Nierensteinen ist der niedrige pH-Wert im Urin der Betroffenen.[110][2]
Je nach Autor und durchgeführter Studie haben 25 bis 75 % aller ADPKD-Patienten mit Zystennieren auch Leberzysten.[111][112][113] Die Anzahl der Leberzysten nimmt mit dem Alter der Patienten zu. Frauen sind von größeren und einer höheren Anzahl von Zysten an der Leber betroffen.[114] Durch die Zysten kann die Leber erheblich vergrößert und regelrecht von Zysten durchsetzt sein. Die Funktion der Parenchymzellen ist in den meisten Fällen jedoch nicht beeinträchtigt.[115][116] So sind beispielsweise die Werte der Leberenzyme und des Bilirubin normal. Weiterreichende Komplikationen ergeben sich eher durch die Platzbeanspruchung der teilweise extrem vergrößerten Leber. Möglich sind dabei beispielsweise ein Hochstand des Zwerchfells, eine Verengung einzelner Darmabschnitte, was zu einem erschwerten Nahrungstransport führen kann und die Obstruktion größerer Blutgefäße, wie beispielsweise der Vena cava inferior.[2]
Die ARPKD führt in der Leber zu einer Fibrose, Zirrhose und zu einem erhöhten Druck in der Pfortader (portale Hypertension).[2]
In anderen Organen wie beispielsweise Bauchspeicheldrüse, Milz oder Eierstöcke finden sich Zysten bei Patienten mit polyzystischen Nieren erheblich seltener.[112]
Bereits 1904 wurde ein Zusammenhang zwischen polyzystischen Nieren und zerebralem Aneurysma beschrieben. Die Daten über die Prävalenz schwanken zwischen 4,5 und 22,5 %.[117][118] Ein möglicher Riss (Ruptur) des betroffenen Blutgefäßes ist eine der gefürchtetsten Komplikationen bei Zystennieren und in nahezu 50 % der Fälle tödlich.[2]
Zystennieren in der Veterinärmedizin
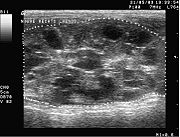 Eine Zystenniere bei einer 13 Jahre alten Katze im Ultraschall
Eine Zystenniere bei einer 13 Jahre alten Katze im Ultraschall→ Hauptartikel: PKD der Katze
Auch in der Tiermedizin sind Zystennieren weit verbreitet. Insbesondere bei Perserkatzen ist dieses Krankheitsbild häufig anzutreffen. Bei ihnen werden Zystennieren autosomal-dominant vererbt. Die Ursache sind – wie in den meisten Fällen beim Menschen – Mutationen des PKD1-Gens. Dieses befindet sich bei Katzen auf dem Chromosom E3. Bei betroffenen Perserkatzen liegt eine Punktmutation in Exon 29 des PKD1-Gens vor.[119][73]
Geschichtliches
 William Osler (1881)
William Osler (1881)Felix Lejars (1863–1932) benutzte in seiner Dissertation[120] 1888 erstmals den Begriff polyzystische Nieren.[121] Der kanadische Mediziner William Osler beschrieb sie 1915.[122][123] Bis zur Mitte des 20. Jahrhunderts befassten sich nur wenige Veröffentlichungen mit diesem Krankheitsbild. Dalgaard erkannte 1957 in seiner Dissertation als erster den autosomal-dominanten Erbgang der ADPKD.[124] 1985 wurde von Reeders und Kollegen der Genlocus von PKD1 auf Chromosom 16 beim Menschen entdeckt.[125]
Einzelnachweise
- ↑ S. Wang u. a.: The autosomal recessive polycystic kidney disease protein is localized to primary cilia, with concentration in the basal body area. In: J Am Soc Nephrol 15, 2004, S. 592–602. PMID 14978161
- ↑ a b c d e f g h i j k l m n o p q r s t U. Faber: Langzeitverlauf bei Adulter Polyzystischer Nierendegeneration nach Nierentransplantation. Dissertation, Heinrich-Heine-Universität Düsseldorf, 2000.
- ↑ J. Milutinovic u. a.: Autosomal dominant polycystic kidney disease: symptoms and clinical findings. In: Q. J. Med. 53, 1984, S. 511–522. PMID 6240069
- ↑ L. W. Elzinga u. a.: Surgical management of painful polycystic kidneys. In: Am. J. Kidney Dis. 22, 1993, S. 532–527. PMID 8213792
- ↑ L. W. Elzinga u. a.: Surgery in the management of autosomal dominant polycystic kidney disease. In: Am. J. Kidney Dis. 1992, S. 89–92. PMID 1739090
- ↑ D. Frang u. a.: A new approach to the treatment of polycystic kidneys. In: Int. Urol. Nephrol. 20, 1988, S. 13–21. PMID 3360583
- ↑ L. W. Elzinga u. a.: Cyst decompression surgery for autosomal dominant polycystic kidney disease. In: J Am Soc Nephrol 2, 1992, S. 1219-1226. PMID 1591362
- ↑ B. J. Lifson u. a.: Role and long-term results of laparoscopic decortication in solitary cystic and autosomal dominant polycystic kidney disease. In: J. Urol. 159, 1998, S. 702–705. PMID 9474129
- ↑ A. B. Chapman u. a.: Overt proteinuria and microalbuminuria in autosomal dominant polycystic kidney disease. In: J Am Soc Nephrol 5, 1994, S. 1349–1354. PMID 7894001
- ↑ P. A. Gabow u. a.: Renal structure and hypertension in autosomal dominant polycystic kidney disease. In: Kidney international 38, 1990, S. 1177–1180. PMID 2074659
- ↑ a b P. A. Gabow: Autosomal dominant polycystic kidney disease In: NEJM 329, 1993, S. 332–342. PMID 8321262
- ↑ A. B. Chapman und P. A. Gabow: Hypertension in autosomal dominant polycystic kidney disease. In: Kidney Int Suppl 61, 1997, S. 71–73. PMID 9328971
- ↑ P. A. Gabow u. a.: Utility of ultrasonography in the diagnosis of autosomal dominant polycystic kidney disease in children. In: J Am Soc Nephrol 8, 1997, S. 105–110. PMID 9013454
- ↑ Polycystic Kidney Disease: MRI Provides An Early Alert To Progression. In: Science Daily vom 17. Mai 2006
- ↑ W. C. O'Neill u. a.: Sonographic assessment of the severity and progression of autosomal dominant polycystic kidney disease: the Consortium of Renal Imaging Studies in Polycystic Kidney Disease (CRISP). In: Am J Kidney Dis 46, 2005, S. 1058–1064. PMID 16310571
- ↑ K. M. Koch u. a.: Klinische Nephrologie. Urban&Fischer-Verlag, 1999, S. 437–459. ISBN 978-3-437-21730-2
- ↑ V. Osathanondh und E. L. Potter: Pathogenesis of polycystic kidneys. In: Arch. Path. 77, 1964, S. 459–465. PMID 14120681
- ↑ B. Hermanns u. a.: Pathologie und Genetik erblicher Zystennieren. In: Der Pathologe, 24, 2003, S. 410–420. PMID 14605845
- ↑ ADPKD (Autosomal Dominante Polyzystische Nierenerkrankung) Universitätsklinikum Aachen, eingesehen am 30. September 2008
- ↑ a b c S. Helmig: Populationsgenetische Untersuchung an dem PKD 1 Gen der Katze im Hinblick auf das Polyzystische Syndrom. Dissertation, Justus-Liebig-Universität Gießen, 2005.
- ↑ F. Hildebrandt und M. Wolf: Pathologie und Genetik hereditärer Zystennieren. In: Medizinische Therapie Springer, 2005, S. 927–939. ISBN 978-3-540-21226-3
- ↑ a b c d e f g W. Kühn und G. Walz: Autosomal dominante polyzystische Nierenerkrankung. In: Ärzteblatt 104/2007, A 3022-8
- ↑ R. G. Elles u. a.: Diagnosis of adult polycystic kidney disease by genetic markers and ultrasonographic imaging in a voluntary family register. In: J Med Genet 31, 1994, S. 115–120. PMID 8182715
- ↑ L. F. Fried u. a.: Duodenal obstruction in polycystic kidney disease. Case report and review of the literature. In: Am. J. Nephrol. 18, 1998, S. 318–320. PMID 9653836
- ↑ a b P. D. Wilson: Polycystic kidney disease. In: N Engl J Med 350, 2004, S. 151–164. PMID 14711914
- ↑ U. Frei und H. J. Schober-Halstenberg: Nierenersatztherapie in Deutschland. In: QuaSi-Niere Jahresbericht 2005/2006 Berlin, Deutschland
- ↑ G. M. Fick und P. A. Gabow: Hereditary and acquired cystic disease of the kidney. In: Kidney Int 46, 1994, S. 951-964. PMID 7861721
- ↑ R. Rohatgi: Clinical manifestations of hereditary cystic kidney disease. In: Front Biosci 13, 2008, S. 4175–4197. PMID 18508505
- ↑ orpha.net: Nierenkrankheit, medulläre zystische, autosomal-dominante, mit oder ohne Hyperurikämie eingesehen am 4. Oktober 2008
- ↑ M. Attanasio u. a.: Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. In: Nature Genet 39, 2007, S. 1018–1024. PMID 17618285
- ↑ a b B. Buchholz: Funktionelle Interaktion von Polycystin 2 und TRPV4. Dissertation, Albert-Ludwigs-Universität Freiburg, 2004.
- ↑ A. R. Gallagher u. a.: Molecular basis of autosomal-dominant polycystic kidney disease. In: Cellular and Molecular Life Sciences 59, 2002, S. 682–693. PMID 12022474
- ↑ Universitätsklinikum Aachen: ADPKD (Autosomal Dominante Polyzystische Nierenerkrankung) eingesehen am 11. November 2008
- ↑ D. W. Bianchi u. a.: Fetology. McGraw-Hill Professional, 2000, ISBN 0-838-52570-9 S. 632.
- ↑ P. A. Gabow u. a.: Factors affecting the progression of renal disease in autosomal dominant polycystic kidney disease. In: Kidney Int 41, 1992, S. 1311–1319. PMID 1614046
- ↑ A. C. Ong und P. C. Harris: Molecular pathogenesis of ADPKD: the polycystin complex gets complex. In: Kidney Int 67, 2005, S. 1234–1247. PMID 15780076
- ↑ S. Rossetti u. a.: Mutation analysis of the entire PKD1 gene: genetic and diagnostic implications. In: Am. J. Hum. Genet. 68, 2001, S. 46-63. PMID 11115377
- ↑ G. G. Germino: Autosomal dominant polycystic kidney disease: a two-hit model. In: Hosp Pract 32, 1997 S. 81-2, 85-8, 91-2. PMID 9078975
- ↑ Y. Pei u. a.: [http://jasn.asnjournals.org/cgi/content/full/10/7/1524 Somatic PKD2 mutations in individual kidney and liver cysts support a "two-hit" model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. In: J Am Soc Nephrol 10, 1999, S. 1524-1529. PMID 10405208
- ↑ F. Qian u. a.: The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. In: Cell 87, 1996, S. 979-987. PMID 8978603
- ↑ T. Watnick u. a.: Mutations of PKD1 in ADPKD2 cysts suggest a pathogenic effect of trans-heterozygous mutations. In: Nature Genetics 25, 2000, S. 143-144. PMID 10835625
- ↑ K. Hackmann: Untersuchungen zur Expression der murinen Gene Pkd1 und Pkd2, den orthologen Genen der Autosomal Dominanten Polyzystischen Nierenerkrankung (ADPKD). Dissertation, Universität Bielefeld, 2005.
- ↑ W. Lu u. a.: Late onset of renal and hepatic cysts in Pkd1-targeted heterozygotes. In: Nature Genetics 21, 1999, S. 160–161. PMID 9988265
- ↑ G. Wu u. a.: Somatic inactivation of Pkd2 results in polycystic kidney disease. In: Cell 93, 1998, S. 177–188. PMID 9568711
- ↑ N. Bogdanova u. a.: Genetic heterogeneity of polycystic kidney disease in Bulgaria. In: Hum Genet 95, 1995, S. 645–650. PMID 7789949
- ↑ M . C. Daoust u. a.: Evidence for a third genetic locus for autosomal dominant polycystic kidney disease. In: Genomics 25, 1995, S. 733–736. PMID 7759112
- ↑ A. D. Paterson und Y. Pei: Is there a third gene for autosomal dominant polycystic kidney disease? In: Kidney International 54, 1998, S. 1759–1761. PMID 9844156
- ↑ M. Koptides und C. C. Deltas: Autosomal dominant polycystic kidney disease: molecular genetics and molecular pathogenesis. In: Hum Genet 107, 2000, S. 115–126. PMID 11030408
- ↑ M. Consugar u. a.: PKD3 revisited with improved PKD1 and PKD2 haplotyping and mutation screening. In: J Am Soc Nephrol 16, 2005, S. 358A.
- ↑ A. D. Paterson und Y. Pei: PKD3-to be or not to be? In: Nephrol Dial Transplant 14, 1999, S. 631–614. PMID 10570111
- ↑ Y. Pei u. a.: Bilineal disease and trans-heterozygotes in autosomal dominant polycystic kidney disease. In: Am J Hum Genet 68, 2001, S. 355-363. PMID 11156533
- ↑ a b S. Rosetti und P. C. Harris: Genotype–Phenotype Correlations in Autosomal Dominant and Autosomal Recessive Polycystic Kidney Disease. In: J Am Soc Nephrol 18, 2007, S. 1374–1380. PMID 17429049
- ↑ a b C. Boucher und R. Sandford: Autosomal dominant polycystic kidney disease (ADPKD, MIM 173900, PKD1 and PKD2 genes, protein products known as polycystin-1 and polycystin-2). In: Eur J Hum Genet 12, 2004, S. 347–354. PMID 14872199
- ↑ C. Stayner und J. Zhou: Polycystin channels and kidney disease. In: Trends in Pharmacological Sciences 22, 2001, S. 543–546. PMID 11698076
- ↑ T. Watnick und G. Germino: From cilia to cyst. In: Nature Genetics 34, 2003, S. 355–356. PMID 12923538
- ↑ B. K. Yoder: Role of Primary Cilia in the Pathogenesis of Polycystic Kidney Disease. In: J Am Soc Nephrol 18, 2007, S. 1381–1388. PMID 17429051
- ↑ B. K. Yoder u. a.: Molecular pathogenesis of autosomal dominant polycystic kidney disease. In: Expert Rev Mol Med 17, 2006, S. 1-22. PMID 16515728
- ↑ K. D. Gardner u. a.: Why renal cysts grow. In: Am J Physiol 266, 1994, F353-359. PMID 8160782
- ↑ D. Rizk und A. B. Chapman: Cystic and inherited kidney diseases. In: Am J Kidney Dis 42, 2003, S. 1305–1317. PMID 14655206
- ↑ M. H. K. Shokeir: Expression of adult polycystic kidney disease in childhood: a longitudinal study. In: Clin. Genet. 14, 1978, S. 61–72. PMID 688689
- ↑ N. Gretz u. a.: Is gender a determinant for evolution of renal failure? A study in autosomal dominant polycystic kidney disease. In: Am J Kidney Dis 14, 1989, S. 178–183. PMID 2672797
- ↑ R. Sherstha u. a.: Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease. In: Hepatology 26, 1997, S. 1282–1286. PMID 9362373
- ↑ P. C. Harris u. a.: Cyst number but not the rate of cystic growth is associated with the mutated gene in ADPKD. In: J Am Soc Nephrol 17, 2006, S. 3013–3019. PMID 17035604
- ↑ R. Magistroni u. a.: Genotyperenal function correlation in type 2 autosomal dominant polycystic kidney disease. In: J Am Soc Nephrol 14, 2003, S. 1164–1174. PMID
- ↑ S. R. Orth u. a.: Smoking as a risk factor for end-stage renal failure in men with primary renal disease. In: Kidney Int 54, 1998, S. 926–931. PMID 9734618
- ↑ S. R. Orth u. a.: Smoking as a risk factor for end-stage renal failure in patients with primary renal disease. In: Contrib Nephrol 130, 2000, S. 109–123. PMID 10892557
- ↑ a b N. Hateboer: Comparison of phenotypes of polycystic kidney disease types 1 and 2. In: The Lancet 353, 1999, S. 103–107. PMID 10023895
- ↑ G. M. Fick u. a.: Causes of death in autosomal dominant polycystic kidney disease. In: J Am Soc Nephrol 5, 1995, S. 2048-2456. PMID 7579053
- ↑ Mayo Medical Clinic: Autosomal Recessive Polycystic Kidney Disease (ARPKD), Known Mutation. eingesehen am 11. November 2008
- ↑ R. Witzgall: New Developments in the Field of Cystic Kidney Diseases. In: Current Molecular Medicine 5, 2005, S. 455–465. PMID 16101475
- ↑ K. Zerres u.a.: Autosomal recessive polycystic kidney disease. In: J Mol Med 76, 1998, S. 303–309. PMID 9587064
- ↑ S. T. Shaikewitz u. a.: Autosomal recessive polycystic kidney disease: Issues regarding the variability of clinical presentation. In: J Am Soc Nephrol 3, 1993, S. 1858–1862. PMID 8338916
- ↑ a b E. C. Kappe: Molekularbiologische Untersuchungen am PKD1-Gen der Katze. Dissertation, Justus-Liebig-Universität Giessen, 2008.
- ↑ M. Z. Zhang u. a.: PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells. In: Proc Nat Acad Sci 101, 2004, S. 2311-2316. PMID 14983006
- ↑ L. M. Guay-Woodford: Renal cystic diseases: diverse phenotypes converge on the cilium/centrosome complex. In: Pediatric Nephrology 21, 2006, S. 1369–1376. PMID 16823577
- ↑ T. Benzing u. a.: Wnt signaling in polycystic kidney disease. In: J Am Soc Nephrol 18, 2007, S. 1389-1398. PMID 17429050
- ↑ S. J. Ansley u. a.: Basal body dysfunction is a likely cause of pleiotropic Bardet–Biedl syndrome. In: Nature 425, 2003, S. 628–633. PMID: 14520415
- ↑ Sampson et al.: Renal cystic disease in tuberous sclerosis: role of the polycystic kidney disease 1 gene. Am J Hum Genet. 1997 Oct;61(4):843-51. PMID 9382094
- ↑ A. A. Connacher u. a.: Orofaciodigital syndrome type I associated with polycystic kidneys and agenesis of the corpus callosum. In: J. Med. Genet. 24, 1987, S. 116–122. PMID 3560170
- ↑ S. A. Feather u. a.: Oral-facial-digital syndrome type 1 is another dominant polycystic kidney disease: clinical, radiological and histopathological features of a new kindred. In: Nephrol. Dial. Transplant 12, 1997, S. 1354–1361. PMID 9249769
- ↑ E. Prati: Oro-facio-digital sydrome type 1. In: Orphanet Encyclopedia Oktober 2004
- ↑ K. Zerres und S. Rudnik-Schöneborn: Polyzystische Nierenerkrankungen. In: Handbuch der Molekularen Medizin Springer-Verlag, Band 7 (Teil 2), 2000, S. 281–295.
- ↑ a b M. A. Matson und E. P. Cohen: Acquired cystic kidney disease: occurrence, prevalence, and renal cancers. In: Medicine (Baltimore) 69, 1990, S. 217–226. PMID 2374506
- ↑ W. Remmele: Pathologie 5. Springer, 1997, ISBN 3-540-61098-7 S. 172.
- ↑ Universität Basel:Sekundäre Zystenniere nach Dialyse wegen Analgetika-Nephropathie. Abbildung eines Histologischen Präparates, eingesehen am 8. September 2008
- ↑ F. A. Belibi u. a.: The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. In: J Am Soc Nephrol 13, 2002, S. 2723–2729. PMID 12397042
- ↑ M. Schmid u. a.: Natriuresis-pressure relationship in polycystic kidney disease. In: J. Hypertens. 8, 1990, S. 277–283. PMID 2159509
- ↑ B. Z. Colleen: Polycystic Kidney Disease: An Overview and Commentary. In: Dialysis and Transplantation 28, 1999, S. 468–474.
- ↑ H. H. Knispel u. a.: Transplantation in autosomal dominant polycystic kidney disease without nephrectomy. In: Urol. Int. 56, 1996, S. 75–78. PMID 8659014
- ↑ Y. Pirson u. a.: Outcome of renal replacement therapy im autosomal dominant polycystic kidney disease. In: Nephrol Dial Transplant 11, 1996, S. 24–28. PMID 9044324
- ↑ J. J.Grantham: Polycystic kidney disease: old disease in a new context. In: Trans Am Clin Climatol Assoc 113, 2002, S. 211–224. PMID 12053711
- ↑ Calvet JP.: Strategies to inhibit cyst formation in ADPKD. In.: Clin J Am Soc Nephrol. 2008 Jul;3(4):1205-11. PMID 18434615
- ↑ Chapman AB.: Approaches to testing new treatments in autosomal dominant polycystic kidney disease: insights from the CRISP and HALT-PKD studies. In: Clin J Am Soc Nephrol. 2008 Jul;3(4):1197-204. PMID 18579674
- ↑ Grantham JJ et al: Volume progression in polycystic kidney disease. In.: N Engl J Med. 2006 May 18;354(20):2122-30 PMID 16707749
- ↑ J. M. Shillingford u. a.: The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. In: Proc Natl Acad Sci 103, 2006, S. 5466–5471. PMID 16567633
- ↑ Y. Tao u. a.: Rapamycin Markedly Slows Disease Progression in a Rat Model of Polycystic Kidney Disease. In: J Am Soc Nephrol 16, 2005, S. 46–51. PMID 15563559
- ↑ P. R. Wahl u. a.: Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). In: Nephrol Dial Transplant 21, 2006, S. 598–604. PMID 16221708
- ↑ S. M. Flechner u. a.: Transplantation. 74, 2002, S. 1070–1076. PMID 12438948
- ↑ Q. Qian u. a.: Sirolimus reduces polycystic liver volume in ADPKD patients. In: J Am Soc Nephrol 19, 2008, S. 631–638. PMID 18199797
- ↑ Edelstein CL.: Mammalian target of rapamycin and caspase inhibitors in polycystic kidney disease. Clin J Am Soc Nephrol. 2008 Jul;3(4):1219-26. PMID 18587045
- ↑ a b A. Masoumi u. a.: Potential pharmacological interventions in polycystic kidney disease. In: Drugs 67, 2007, S. 2495–2510. PMID 18034588
- ↑ F. A. Belibi u. a.: Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. In: Kidney Int 66, 2004, S. 964–973. PMID 15327388
- ↑ T. Yamaguchi u. a.: cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. In: Kidney Int 57, 2000, S. 1460–1471. PMID 10760082
- ↑ Triptolide: A Potential Drug For Polycystic Kidney Disease. In: Science Daily vom 12. März 2007
- ↑ Experiments Point To New Treatments For PKD. In: Science Daily vom 6. April 2008
- ↑ S. J. Leuenroth u. a.: Triptolide Reduces Cystogenesis in a Model of ADPKD. In: J Am Soc Nephrol 19, 2008, S. 1659–1662. PMID 18650476
- ↑ S. J. Leuenroth u. a.: Triptolide is a traditional Chinese medicine-derived inhibitor of polycystic kidney disease. In: Proc Natl Acad Sci 104, 2007, S. 4389–4394. PMID 17360534
- ↑ Torres VE.: Role of vasopressin antagonists. In: Clin J Am Soc Nephrol. 2008 Jul;3(4):1212-8. PMID 18434616
- ↑ Torres VE. et al.: Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. In.: Nat Med 2004 Apr;10(4):363-4. PMID 14991049
- ↑ V. E. Torres u. a.: Renal stone disease in autosomal dominant polycystic kidney disease. In: Am. J. Kidney Dis. 22, 1993, S, 513–519. PMID 8213789
- ↑ J. Milutinovic u. a.: Liver cysts in patients with autosomal dominant polycystic kidney disease. In: Am. J. Med. 68, 1980, S. 741–744. PMID 7377224
- ↑ a b E. Higashihara u. a.: Clinical aspects of polycystic kidney disease. In: J. Urol. 147, 1992, S. 329–332. PMID 1732586
- ↑ Y. Itai u. a.: Hepatobiliary cysts in patients with autosomal dominant polycystic kidney disease: prevalence and CT findings. In: Am. J. Roentgenol. 164, 1995, S. 339–342. PMID 7839965
- ↑ P. A. Gabow u. a.: Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. In: Hepatology 11, 1990, S. 1033–1037. PMID 2365280
- ↑ E. Levine u. a.: Liver cysts in autosomal-dominant polycystic kidney disease: clinical and computed tomographic study. In: Am. J. Roentgenol. 145, 1985, S. 229–233. PMID 3875218
- ↑ A. Telenti u. a.: Hepatic cyst infection in autosomal dominant polycystic kidney disease. In: Mayo Clin. Proc. 65, 1990, S. 933–942. PMID 2198396
- ↑ W. I. Schievink u. a.: Saccular intracranial aneurysms in autosomal dominant polycystic kidney disease. In: J. Am. Soc. Nephrol. 3, 1992, S. 88–95. PMID 1391712
- ↑ A. B. Chapman u. a.: Intracranial aneurysms in autosomal dominant polycystic kidney disease. In: NEJM 327, 1992, S. 916–920. PMID 1513348
- ↑ L. A. Lyons u. a.: Feline Polycystic Kidney Disease mutation identified in PKD1. In: J. Am. Soc. Nephrol. 15, 2004, S. 2548–2555. PMID 15466259
- ↑ F. Lejars: Du gros reins polykystique de l'adulte. Dissertation, 1888, Paris
- ↑ B. Schulze: Zystennieren: Auf dem Weg zu einer behandelbaren Erkrankung. In: MedReport 44, 2006, S. 5.
- ↑ W. Osler: The diagnosis of polycystic kidney. In: Internat Clin Philadelphia, 2, 1915, S. 1–5.
- ↑ L. P. Brendan u. a.: Did Sir William Osler Perform an Autopsy at The Johns Hopkins Hospital? In: Archives of Pathology and Laboratory Medicine 2, 132, 2007, S. 261–264.
- ↑ O. Z. Dalgaard: Bilateral polycystic disease of the kidneys. In: Acta Med Scand 328, 1957, S. 1–255. PMID 13469269
- ↑ S. T. Reeders u. a.: A highly polymorphic DNA marker linked to adult polycystic kidney disease on chromosome 16. In: Nature 317, 1985, S. 542–544. PMID 2995836
Literatur
Fachartikel
- W. Kühn und G. Walz: Autosomal dominante polyzystische Nierenerkrankung. In: Dtsch Arztebl 104, 2007, S. A3022–8.
- S. Shab-Par: Polyzystische Nierenerkrankung: Everolimus im Probelauf. In: ClinCum 10, 2007.
- I. Ishikawa: Acquired renal cystic disease. In: The Cystic Kidney Kluwer, 1990, ISBN 9-780-79230-3923 S. 351–377.
- J. J. Grantham und P. A. Gabow: Polycystic Kidney Disease. In: Diseases of the Kidney Little Brown, 1988, S. 583–615.
Fachbücher
- M. L. Watson (Editor): Polycystic kidney disease. Oxford Univ. Press, 1996. ISBN 0-192-62578-0
- H. M. Sass und P. Schröder (Herausgeber): Patientenaufklärung bei genetischem Risiko. LIT Verlag, 2003, S. 147–198. ISBN 3-825-84987-2
Patienteninformationen
- A. B. Chapman und L. M. Guay-Woodford: The Family and ADPKD: A Guide for Children and Parents. Polycystic Kidney Research Foundation, 1997, ISBN 0-961-45675-2
Populärwissenschaftlich
- T. Kotlorz: Neue Hoffnung für Nierenkranke. In: Die Welt Ausgabe vom 23. Juli 2007
- H. Jänz: Hoffen auf die Niere. In: Die Welt Ausgabe vom 3. Juni 2006
Weblinks
- PKD Familiäre Zystennieren e. V.
- Urologielehrbuch: Autosomal dominante polyzystische Nierenerkrankung (ADPKD)
- zystennieren.de Ruhr-Universität Bochum

Bitte beachte den Hinweis zu Gesundheitsthemen!












Wikimedia Foundation.