- Blosum
-
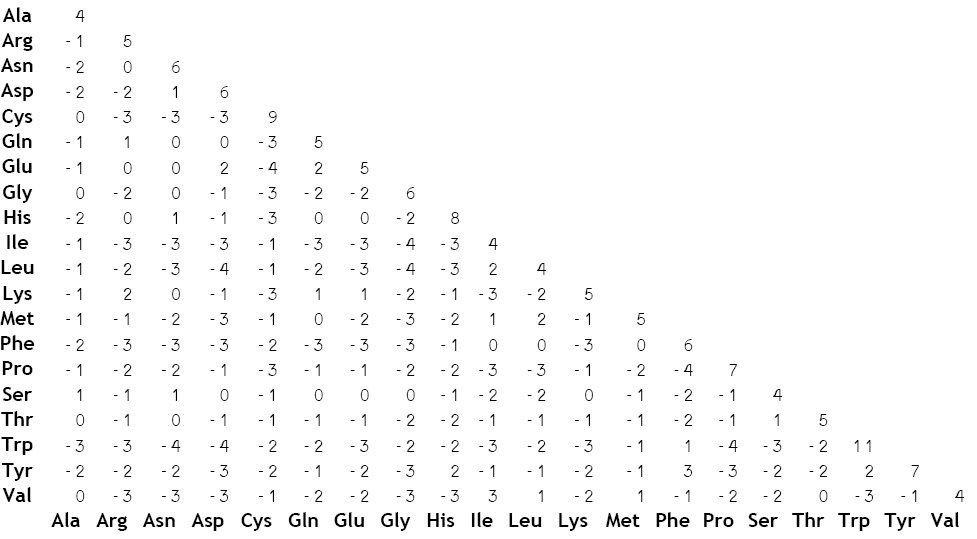 Die BLOSUM62-Matrix
Die BLOSUM62-MatrixBLOSUM (BLOcks SUbstitution Matrix[1]) ist eine evidenzbasierte Substitutionsmatrix, die für Sequenzalignment von Proteinen benutzt wird und spielt neben der Point Accepted Mutation Matrix (PAM-Matrix) eine wichtige Rolle in der Bioinformatik. Die BLOSUM wurde 1992 von Jorja G. Henikoff und Steven Henikoff entwickelt. Es existieren verschiedene Matrizen für unterschiedliche evolutionäre Distanzen. [2]
Inhaltsverzeichnis
Berechnung
BLOSUM verwendet einzelne Blöcke (ohne Lücken) von homologen Proteinen, die verglichen werden. Es existieren verschiedene BLOSUM, welche für verschiedene Anwendungsbereiche konzipiert sind. So sind BLOSUM mit hohen Nummern wie BLOSUM80 für evolutionär nahe verwandte Proteine geeignet und solche mit tiefen Nummern wie BLOSUM45 für stark divergierende Proteine geeignet. Die Autoren der BLOSUM haben entsprechend der Matrixnummer alle Blöcke, die eine höhere Sequenzidentität als die festgelegte Prozentzahl aufwiesen, zu einer einzigen Sequenz zusammengefügt (clustering), um damit den Einfluss von nahe verwandten Blöcken zu mindern. So wurden für BLOSUM80 alle Sequenzen mit mehr als 80% Sequenzidentität zusammengefügt, so dass alle verbleibenden Sequenzen untereinander verglichen weniger als 80% Identität hatten.[2] In die Matrix eingetragen werden die log-odds Werte:
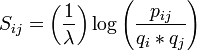
wobei pi,j die Wahrscheinlichkeit die Aminosäuren i und j in einem Alignment zu finden und qi bzw. qj die Häufigkeit der Aminosäuren überhaupt bezeichnet. λ ist ein Normalisierungsfaktor, die Werte werden auf ganze Zahlen gerundet. Somit ist der Logarithmus größer als Null und eine positive Score resultiert, wenn die beiden Aminosäuren häufiger in einem Alignment gefunden werden als nur durch Zufall erwartet würde. So ist z.B. der Wert für eine Substitution von Tryptophan mit Tyrosin in der BLOSUM62 mit 2 größer als Null, was bedeutet, dass Tryptophan zu Tyrosin (und umgekehrt) häufiger mutiert als nur durch Zufall zu erwarten wäre - dies macht auch aufgrund der ähnlichen physikalischen und chemischen Eigenschaften der beiden Aminosäuren Sinn. Die größte Score wird jedoch meist für die Identität beobachtet, so hat ein Tryptophan, das ein Tryptophan bleibt eine Score von 11 und ein Tyrosin, das ein Tyrosin bleibt, eine Score von 7.
Der Vorteil der log-odds ist, dass diese addiert werden können anstatt multipliziert wie normalerweise Wahrscheinlichkeiten und dies die Berechnung numerisch einfacher macht. Die Wahrscheinlichkeit selber kann einfach zurückgewonnen werden, indem die Score exponenziert wird.Verwendung
BLOSUM mit hohen Zahlen (z.B. BLOSUM80) werden für den Vergleich von nahe verwandten Sequenzen verwendet während BLOSUM mit tiefen Zahlen für den Vergleich von entfernt verwandten Proteinen verwendet wird. Oftmals wird ein Alignment von zwei Sequenzen durch die Verwendung der BLOSUM evaluiert. So hat z.B. das folgende Alignment
EKNGFPA
| | |
EMQGRWAmit der BLOSUM62 die Score 7.
Die Algorithmen, welche entweder globales (Needleman & Wunsch) oder lokales (Smith & Waterman) paarweises Sequenzalignment durchführen, verwenden für Proteinsequenzen oft BLOSUM als Substitutionsmatrix, allerdings kann dies frei gewählt werden. Die Algorithmen BLAST oder FASTA, welche eine Datenbank nach einer bestimmten Sequenz durchsuchen, verwenden für Proteinsuchen auch häufig BLOSUM. Dabei ist der Benutzer oftmals nicht in exakten Treffern interessiert und wenn auch verwandte aber nicht identische Proteine gesucht werden, dann kann mithilfe der BLOSUM evaluiert werden, ob das Alignment zu einem bestimmten Protein in der Datenbank signifikant ist oder nicht.Einzelnachweise
- ↑ Im Akronym BLOSUM steht das letzte 'M' bereits für 'Matrix' und deshalb ist es falsch, von einer 'BLOSUM matrix' zu sprechen, da dies ein redundantes Akronym ist.
- ↑ a b Henikoff S, Henikoff JG: Amino acid substitution matrices from protein blocks. Proceedings of the National Academy of Sciences USA. 1992 Nov 15;89(22):10915-9. PMID 1438297
Weblinks
Siehe auch
Wikimedia Foundation.