- CLL
-
Klassifikation nach ICD-10 C91.1 Chronische lymphatische Leukämie
ICD-O 9823/3 (CLL)
ICD-O 9670/3 (B-SLL)ICD-10 online (WHO-Version 2006) Die chronische lymphatische Leukämie (CLL) ist ein niedrigmalignes, leukämisch verlaufendes B-Zell-Non-Hodgkin-Lymphom ("B-NHL"). Sie ist in der westlichen Welt die am häufigsten vorkommende Leukämieform und tritt vor allem im höheren Lebensalter auf (das Durchschnittsalter bei Diagnosestellung liegt bei über 50 Jahren). Die WHO-Klassifikation der hämatologischen Erkrankungen unterscheidet neben der CLL noch eine Unterform, das "small lymphocytic lymphoma" (B-SLL, Kleinzelliges B-Zell-Lymphom), das im wesentlichen einer CLL entspricht, bei der der Lymphknotenbefall ganz im Vordergrund steht, ohne dass es zu einer höhergradigen Manifestation im Blut ("Leukämie") kommt (gewissermaßen eine nicht-leukämisch verlaufende CLL).
Inhaltsverzeichnis
Epidemiologie
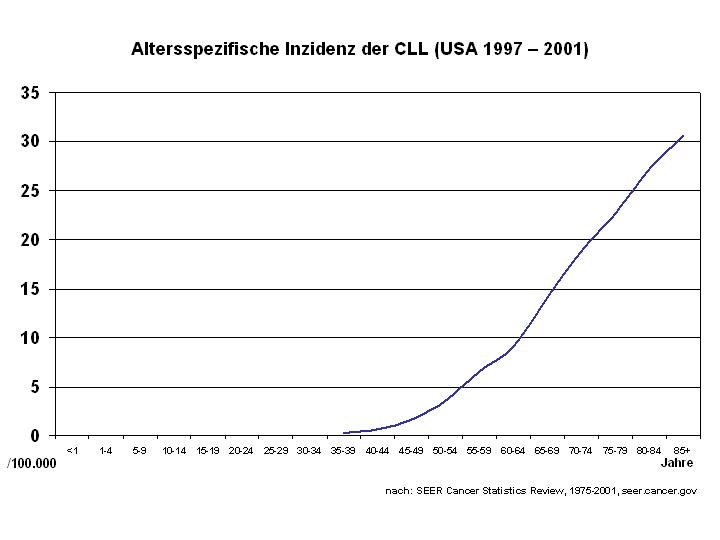
Die Inzidenz (Zahl der Neuerkrankungen) pro Jahr beträgt ca. 3 pro 100.000 Einwohner. Die CLL ist die häufigste Leukämie in der westlichen Welt. Männer erkranken häufiger als Frauen (M : F = 1,7 : 1). Das mediane Alter beträgt bei Erstdiagnose 70 bis 75 Jahre.[1]
Pathogenese
Bei der Erkrankung kommt es zu einer klonalen Vermehrung von reifen, kleinzelligen aber funktionslosen B-Lymphozyten. Die genaue Ursache hierfür ist nicht bekannt. Es kann davon ausgegangen werden, dass genetische Veränderungen, die im Laufe des Lebens erworben wurden, entscheidend sind. Hinweise für eine infektiöse Ursache, z. B. durch Viren, gibt es bisher nicht.
Mit klassischen zytogenetischen Methoden sind häufig keine Chromosomentranslokationen nachzuweisen, wie das bei vielen anderen Non-Hodgkin-Lymphomen der Fall ist. Die genauere molekulare Analyse hat aber viele verschiedene genetische Veränderungen gezeigt. Insgesamt kann heute gesagt werden, dass die CLL, was die genauen molekularen Ursachen angeht, ein heterogenes Krankheitsbild ist. Es gibt Patienten, bei denen die Erkrankung einen sehr gutartigen Verlauf nimmt, aber auch eine kleine Untergruppe von Patienten, bei denen die Erkrankung einen deutlich aggressiveren Verlauf zeigt. Diese Unterschiede sind sicher Folge der unterschiedlichen genetischen Veränderungen bei verschiedenen Patienten.
Diagnose
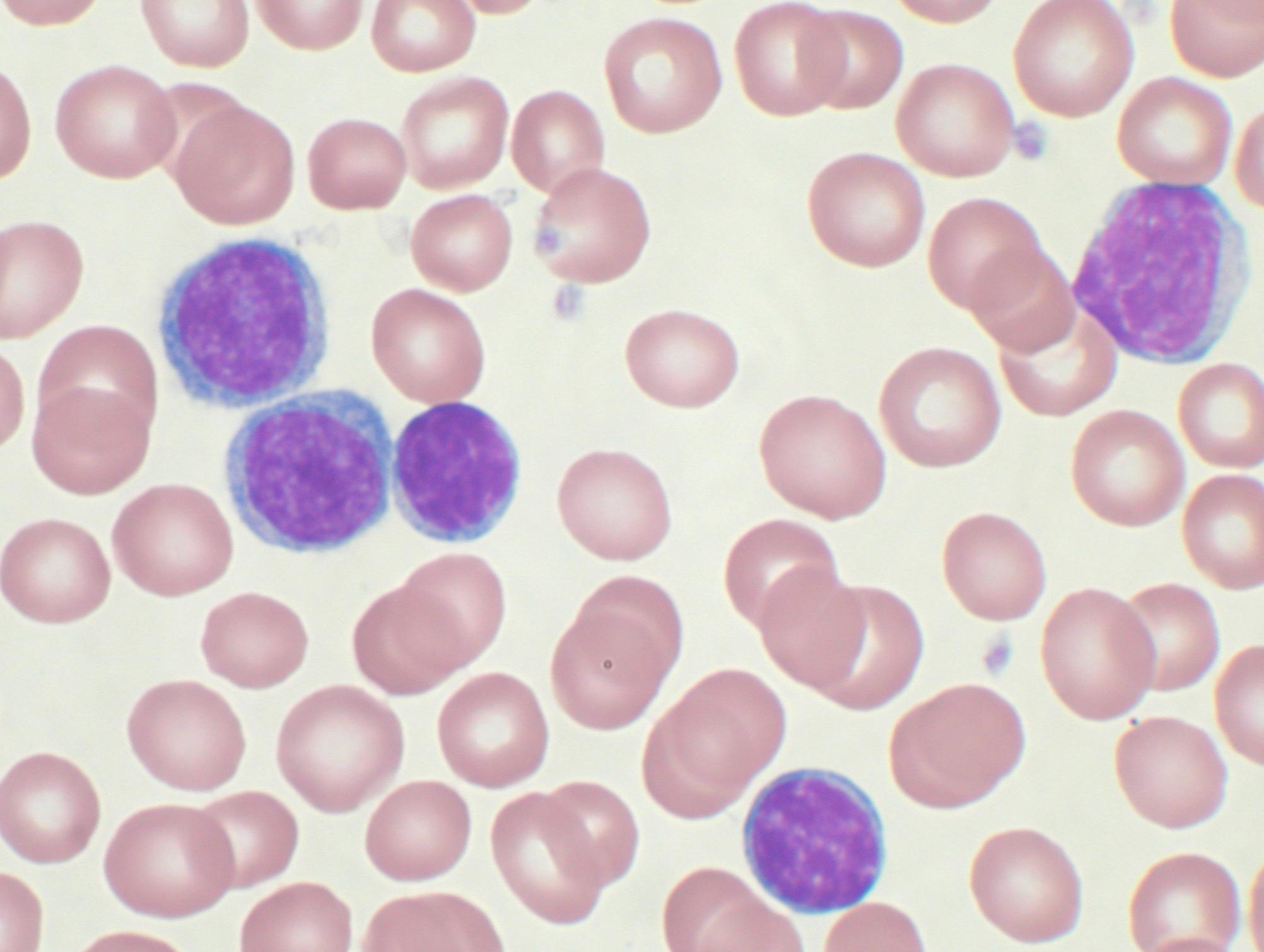
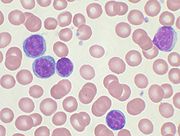 Blutausstrich bei CLL
Blutausstrich bei CLLDie Diagnose der CLL wird in der Regel gestellt durch zwei einfache Untersuchungen:
- Blutbild und Differentialblutbild. Hier wird eine Erhöhung der Lymphozyten auf mindestens 5000/µl für die Dauer von mindestens 4 Wochen für die Diagnosestellung gefordert. Es finden sich die typischen reifzelligen, kleinen Lymphozyten sowie Gumprechtsche Kernschatten (entstehen als Artefakt bei der Präparation) im Blutausstrich.
- Immunphänotypisierung der Leukämiezellen im peripheren Blut. Diese zeigt eine mit den Antikörpern gegen typische B-Zellmarker (CD19) markierbare Zellpopulation, die außerdem CD23 und das T-Zell-Antigen CD5 auf der Oberfläche exprimiert (CD5/CD19-Koexpression), sowie eine Leichtkettenrestriktion (kappa oder lambda).
Weitere Untersuchungen dienen der Erkennung der Ausbreitung der CLL (Röntgen-Thoraxaufnahme, Ultraschalluntersuchung des Abdomen).
Symptome
Häufig ist die Entdeckung der Krankheit ein Zufallsbefund während einer Blutuntersuchung im Rahmen der Diagnostik anderer Erkrankungen. Folgende Symptome treten im Verlauf der Erkrankung auf:
- Lymphknotenschwellungen
- Milz/Lebervergrößerung
- Hauterscheinungen (30 Prozent der Fälle)
- Pruritus
- Ekzeme
- Mykosen
- Herpes zoster
- Hautblutungen
- knotige Infiltrate
- blasse Haut und Schleimhaut
- Parotis/Ohrspeicheldrüsenschwellung ( Mikulicz - Syndrom )
- Leukozytose mit Lymphozytose > 10.000 /µl
- im Knochenmark hoher Anteil reifer Lymphozyten
- Antikörpermangelsyndrom durch Verdrängung der B-Zellen
- Nachweis inkompletter Wärmeautoantikörper
- verminderte Gammaglobuline
- erhöhte IgM
Differentialdiagnose
- Leukämisches Non-Hodgkin-Lymphom
- Haarzellleukämie
- Prolymphozytenleukämie
Therapie
Die Therapie hängt vom jeweiligen Stadium der Erkrankung ab. Die klinische Einteilung nach Binet unterscheidet drei Stadien:
Stadium Befund Untergruppe A Lymphozytose mit Befall von weniger als 3 Lymphknotenregionen an Hals, Achsel, Leiste, Leber oder Milz A(0) keine vergrößerten Knoten
A(I) vergrößerte Knoten
A(II) Leber/MilzvergrößerungB 3 oder mehr der o. g. Lymphknotenregionen befallen B(I) keine vergrößerten Knoten
B(II) Leber/MilzvergrößerungC Anämie (Hb < 10 g/dl) oder Thrombozytopenie (Thrombozyten < 100/nl) C(III) Anämie
C(IV) ThrombozytopenieIm Stadium A(0) wird nur symptomatisch mit Antibiotika / Antimykotika behandelt. Ab Stadium A(I) wird behandelt, wenn folgende Komplikationen auftreten:
- Anämie
- Thrombozytopenie mit Blutungsgefahr
- Milzvergrößerung
- Beschwerden durch wachsende Tumore
Es wird mit den Chemotherapeutika Chlorambucil + Prednison ( Knospe - Schema ) oder Chlorodeoxyadenosin und Deoxyformycin bzw. Fludarabin behandelt. Die Deutsche CLL Studiengruppe empfiehlt derzeit als Standardtherapie zur ersten Behandlung einer CLL eine Kombination aus Fludarabin und Cyclophosphamid. Wahrscheinlich wird demnächst eine zusätzliche Ergänzung mit dem CD20-Antikörper Rituximab empfohlen, jedoch müssen aktuelle Studienergebnisse abgewartet werden. Eine "Evidence based", d. h. "nachgewiesen wirksame" Therapie für Patienten, die nach einer ersten Therapie ein Rezidiv erleiden, gibt es derzeit noch nicht. Bei Patienten, die nicht auf Fludarabin ansprechen, wird der Anti-CD52-Antikörper Alemtuzumab eingesetzt. Des weiteren kommt eine Knochenmark- oder Stammzelltransplantation in Betracht. Allerdings sind die allogenen Tranplantationsstrategien bei der CLL mit hohen therapiebedingten Sterblichkeitsraten verbunden und kommen nur bei ausgewählten Patienten zur Anwendung. Zur Behandlung größerer Lymphome wird die Bestrahlung eingesetzt.
Literatur
- Kurt Possinger und Anne C. Regierer (Hrsg.): Facharzt Hämatologie, Onkologie. München, Jena: Elsevier, Urban & Fischer, 2006. ISBN 3-437-23770-5.
- Hermann Delbrück: Chronische Leukämien: Rat und Hilfe für Betroffene und Angehörige. 2. Auflage. Stuttgart: Kohlhammer, 2004. ISBN 3-17-018369-9.
- Michael Hallek und Bertold Emmerich (Hrsg.): Chronische lymphatische Leukämie. 2. Auflage. Bremen, London, Boston: UNI-MED-Verlag, 2005. ISBN 3-89599-865-6.
- Tait D. Shanafelt, Susan M. Geyer und Neil E. Kay: Prognosis at Diagnosis: Integrating Molecular Biologic Insights into Clinical Practice for Patients with CLL. In: Blood 103 (2004) S. 1202–1210. (PMID 14576043)
Einzelnachweise
- ↑ Kurt Possinger und Anne Regierer (Hrsg.) Facharzt Hämatologie und Onkologie, Verlag Elsevier München, ISBN 978-3437237706
Weblinks

Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.