- Celsentri
-
Strukturformel 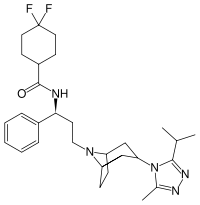
Allgemeines Freiname Maraviroc Andere Namen - (S)-4,4-Difluorcyclohexan carbonsäure- {(1R,3S,5S)-3-[3-(3-isopropyl-5- methyl-[1,2,4]triazol-4-yl) -8-azabicyclo[3.2.1]oct-8-yl] -1-phenylpropyl}amid
- Latein: Maravirocum
Summenformel C29H41F2N5O CAS-Nummer 376348-65-1 PubChem 3002977 ATC-Code J05AX09
DrugBank DB04835 Arzneistoffangaben Wirkstoffklasse Wirkmechanismus CR5-Korezeptor Antagonist
Fertigpräparate - Celsentri® (USA) (D)
Verschreibungspflichtig: Ja Eigenschaften Molare Masse 513,67 g·mol−1 Sicherheitshinweise Gefahrstoffkennzeichnung
unbekanntR- und S-Sätze R: ? S: ? Bitte beachten Sie die eingeschränkte Gültigkeit der Gefahrstoffkennzeichnung bei Arzneimitteln Soweit möglich und gebräuchlich, werden SI-Einheiten verwendet. Wenn nicht anders vermerkt, gelten die angegebenen Daten bei Standardbedingungen. Maraviroc (Handelsname: Celsentri®, Hersteller: Pfizer) ist ein Arzneistoff zur Behandlung von HIV-Infektionen und dessen späteren Stadien[1]. Er gehört zur Gruppe der Entry-Inhibitoren (Corezeptor-Antagonisten).
Inhaltsverzeichnis
Pharmakologie
Maraviroc ist ein Entry-Inhibitor, der als selektiver Inhibitor den menschlichen Chemokinrezeptor CCR5 blockiert und somit das Andocken von HI-Viren an menschliche Zellen, insbesondere Makrophagen verhindert. Die Zelle wird nicht infiziert. Seine Wirkung ist jedoch auf CCR5 nutzende (R5) HI-Viren beschränkt.
Pharmakokinetik
Maraviroc wird aus dem Magendarmtrakt resorbiert. Die empfohlene Dosierung beträgt zweimal täglich 150, 300 oder 600 mg in Abhängigkeit von der übrigen HAART, die durch Interaktion das Verhalten von Maraviroc mehr oder weniger beeinflusst. Die Bioverfügbarkeit liegt bei 23 bis 33%. Die Einnahme des Arzneimittels kann mit oder ohne Nahrung erfolgen. Der Wirkstoff wird zu 76 % an Plasmaproteine gebunden, das Verteilungsvolumen wurde mit 2,8 ± 0,9 l/kg berechnet.
Maraviroc wird über Cytochrom P450 Monooxygenasen verstoffwechselt. Ein relevanter induktorischer oder inhibitorischer Effekt wurde nicht nachgewiesen. Wichtigstes Stoffwechselprodukt ist ein am Stickstoff desalkylierter Metabolitt. Die Substanz wird hauptsächlich über den Stuhl ausgeschieden (ca. 25% unverändert); 8% einer Dosis von 300 mg erscheinen unverändert im Urin. Nach intravenöser Gabe beträgt Eliminationshalbwertzeit ca. 13 Stunden.
Bei Patienten mit angeborener oder medikamentös induzierter Niereninsuffizienz ist unter Umständen eine Dosisanpassung erforderlich (Einbeziehung der übrigen HAART Medikamente). Bei leichter oder mittlerer Einschränkung der Leberfunktion sind die Blutspiegel um ca. 25 bis 46% erhöht. Für diese Patientengruppe liegen noch nicht ausreichend Daten vor. Studien bei Kindern und älteren Patienten wurden noch nicht durchgeführt.
Nebenwirkungen
Husten, Pyrexie, Infektionen der oberen Luftwege, Rash, Beschwerden des Muskelskelettsystems, Abdominalschmerzen und Schwindel sind die am häufigsten berichteten unerwünschten Ereignisse, weiterhin waren Diarrhoe, Oedeme, Influenza, ösophagale Candidiasis, Schlafstörungen, Rhinitis, Parasomnie und Harnwegsabnormalitäten zu verzeichnen.[2]
Quellen
- ↑ AIDS Meds - Amerikanische HIV Medikamenten Website
- ↑ http://www.kompendium.ch/MonographieTxt.aspx?lang=de&MonType=fi
Weblinks

Bitte beachte den Hinweis zu Gesundheitsthemen!

Wikimedia Foundation.