- Chromosom-18p-Monosomie
-
De-Grouchy-Syndrom ist die Bezeichnung für zwei Typen einer chromosomalen Mutation beim Menschen, die auf Verluste (Deletionen) verschiedener Stücke des kurzen (18p-) bzw. langen Arms (18q-) von Chromosom 18 zurückzuführen sind. Es entstehen dabei partielle Monosomien; der kritische Chromosomenabschnitt liegt im Genort 18q23.
Das Syndrom gliedert sich in die beiden Typen De-Grouchy-Syndrom I und De-Grouchy-Syndrom II, die unter dem Oberbegriff Deletions-Syndrome des Chromosoms 18 zusammengefasst werden. Sie gehen jeweils mit verschiedenen Fehlbildungskomplexen einher. Eine Kombination von Merkmalen beider Syndromtypen ist bei Menschen mit dem 18-R-Syndrom feststellbar.
Die Erstbeschreibung unter wissenschaftlichen Aspekten erfolgte in den Jahren 1963 und 1964. Seit dem sind mehr als 100 Fallbeispiele dokumentiert.
Inhaltsverzeichnis
Symptome
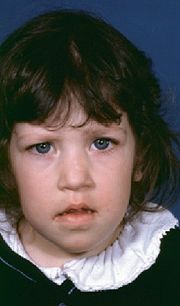 Mädchen mit De-Grouchy-Syndrom
Mädchen mit De-Grouchy-Syndrom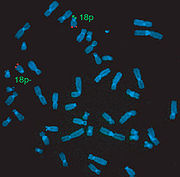 FisH-Test-Ergebnis beim De-Grouchy-Syndrom I
FisH-Test-Ergebnis beim De-Grouchy-Syndrom IDie phänotypische Ausprägung ist sehr unterschiedlich, längst nicht alle Symptome kommen bei allen betroffenen Menschen vor, und sie können individuell verschieden ausgebildet sein. Dies hängt vermutlich (auch) ab von der Länge des jeweils verloren gegangenen Stücks des Chromosoms, obgleich genaue Zusammenhänge noch nicht belegt werden konnten. Rückschlüsse auf die Ausprägung der Symptomatik und die Entwicklung eines betroffenen Kindes sind somit nicht möglich. Sicher spielt die Art und Schwere der individuell vorliegenden Symptome eine wesentliche Rolle für die Prognose. In einigen Fällen kann der Einfluss eines festgestellten Zellmosaiks auf die Ausprägung der Symptomatik nicht ausgeschlossen werden. Bei einem solchen genetischen Mosaik existieren parallel zwei Zelllinien, wobei eine wie üblich und eine auffällig ist (hier mit der entsprechenden Deletion). Abhängig vom Anteil der üblichen Zellen kann die Symptomatik milder ausgeprägt sein.
De-Grouchy-Syndrom I
Der Typ I des Syndroms ist auch unter den Synonymen 18p-Syndrom, 18p-Deletion-Syndrom, Deletion-18p-Syndrom, del(18p)-Syndrom, Chromosom-18p-Monosomie, Monosomie 18p und Partielle Monosomie 18p bekannt. Es liegt ein Stückverlust (Deletion) des kurzen Arms (18p-) von Chromosom 18 vor.
Die Kinder fallen nach der Geburt gehäuft durch ein niedriges Geburtsgewicht und somatische Hypotrophie, verringerte Muskelspannung (Muskelhypotonie), oft recht große Hände mit kurzen Fingern, manchmal Vierfingerfurche, seitliche Biegung eines Fingerglieds (Klinodaktylie) und die Syndaktylie einiger Zehen auf. Herzfehler und Darmlageanomalien (Malrotationen) sind möglich.
Manchmal sind einige dieser und weiterer Symptome nicht sofort offensichtlich, sondern bilden sich erst im Verlauf der ersten Lebensjahre erkennbar aus. Zu den beschriebenen Merkmalen bei Kindern mit dem Typ I des Syndroms zählen vergleichsweise große Ohren, teilweise liegt ein Verschluss (Atresie) oder eine Verengung (Stenose) der Gehörgänge vor, Trichterbrust, Besonderheiten im Bereich des Gehirns und Kopfes (gehäuft Kurzköpfigkeit mit besonderer Rundung des Kopfes / Brachyzephalie, Holoprosenzephalie bei einem von zehn Kindern, teils mit Einäugigkeit / Monophthalmie, Fehlen des Riechhirns / Arrhinenzephalie, vorzeitiger Verschluss der Schädelnähte), Besonderheiten im Bereich der Augen (Fehlbildung der Augenmuskulatur / okulare Dysmorphie, Fehlbildungen der knöchernen Augenhöhlen / orbitare Dysmorphie, vergleichsweise kleine Augen / Mikrophthalmie, recht weit auseinander liegende Augen / Hypertelorismus, ungewöhnlich enge Lidspalten / Blepharophimose mit nach oben oder unten gerichteter Lidachsenstellung, halbmondförmige Hautfalte an den inneren Augenwinkeln / Epikanthus medialis, Herabhängen eines oder beider oberer Augenlider / Ptosis, Katarakt, Glaukom, Kolobom, Schielen / Strabismus, Augenzittern / Nystagmus), ungewöhnlich langer Daumen.
Ebenso beschrieben sind eine deutliche Unterentwicklung (Hypoplasie) und/oder eine Rückverlagerung des Unterkiefers (Distalbiss / mandibuläre Retrognathie), übermäßige Breite der Mundspalte (Makrostomie), herabhängende Mundwinkel, Besonderheiten der Zähne bzw. der Zahnentwicklung (später Zahndurchbruch, teils fehlende Zahnanlagen, Anfälligkeit für Zahnkaries), das Vorliegen einer Verengung (Stenose) oder eines Verschlusses (Atresie) der Choanen, die die Nasenhöhlen mit dem Rachen verbinden, ein retardiertes Knochenalter (dadurch verlangsamtes Wachstum), Wirbelsäulenverbiegung (Skoliose), Buckel (Kyphose). Eine Schilddrüsenentzündung (Thyreoiditis), Morbus Basedow, Immunschwächen mit Autoimmun-Symptomatik, Mangel an Immunglobulin A, im Jugendalter Diabetes mellitus Typ 1 und bei Mädchen das Ausbleiben der Menstruation (primäre Amenorrhoe) können auftreten.
Im Verlauf der Kindesentwicklung zeigt sich eine kognitive Behinderung, deren Grad von leicht bis sehr schwer variieren kann.
De-Grouchy-Syndrom II
Beim Typ II des Syndroms liegt eine Deletion des langen Arms 18q22-23 vor. Auch hier fallen die Kinder durch somatische Hypotrophie und in der weiteren Entwicklung durch hypophysär bedingten Minderwuchs, wahrscheinlich aufgrund einer Wachstumshormoninsuffizienz (GH-Insuffizienz) auf. Mikrozephalie, Muskelhypotonie, Verengung oder Verschluss der Gehörgänge, Augenfehlbildungen, Besonderheiten der Zähne, kognitive Behinderung und Immunschwächen mit Autoimmun-Symptomatik können wie beim Typ I auftreten.
18-R-Syndrom
Liegt bei einem Kind ein Ringchromosom 18 mit Deletion des langen und des kurzen Arms (r18) vor, zeigen sich Kombinationen aus Merkmalen des De-Grouchy-Syndroms I und II.
Diagnose
Auch weil die Deletions-Syndrome des Chromosoms 18 sehr selten und die Symptome variabel und daher schwer zuzuordnen sind, ist eine Chromosomenanalyse das diagnostische Mittel der Wahl. Eine Differentialdiagnose, z. B. zum Cerebro-Okulo-Fazio-Skelettalen Syndrom kann bei der Eingrenzung helfen.
Behandlung
Da es sich um eine chromosomale Ursache handelt, ist eine ursächliche Heilung nicht möglich, so dass sich die Behandlung auf die symptomatische Therapie einzelner Symptome bezieht.
Genetik
Die Deletion besteht meist in einem einfachen Stückverlust am Chromosom 18.
In seltenen Fällen besteht eine Translokation des Chromosoms 18 mit einem anderen Chromosom, die aus einer ursprünglich reziproken und balancierten Form hervorgegangen ist, bei der es zu einem Stückaustausch zwischen zwei Chromosomen oder zu einer perizentrischen Inversion ohne Stückverlust und entsprechend auch ohne phänotypische Auswirkungen gekommen ist. Menschen mit dieser balancierten Chromosomenbesonderheit sind klinisch unauffällig und können sich fortpflanzen. Die Wahrscheinlichkeit, dass es bei der Befruchtung zu einer Inbalance im Erbgut des Kindes kommt, ist möglich, wenn bei Mutter oder Vater eine reziproke Translokation oder eine Inversion vorliegt. Im Verlauf der Reifeteilung kann es bei dieser Konstellation passieren, dass beide Translokationschromosomen weitervererbt werden oder ein sogenanntes crossing over im invertierten Abschnitt erfolgt. Die vormals ausgeglichene Balance des Erbgutes wird somit gestört, und es kommt zum Stückverlust, der beim Kind zum jeweiligen Deletions-Syndrom führt.
Wiederholungswahrscheinlichkeit
In den meisten Fällen tritt die Deletion spontan auf. Ist das Chromosomenbild (Karyotyp) beider Elternteile unauffällig oder hat das betroffene Kind ein Ringchromosomen 18, gibt es derzeit keine Anhaltspunkte für eine Erhöhung der Wahrscheinlichkeit, noch mal ein Kind mit einem der Deletions-Syndrome des Chromosoms 18 zu zeugen.
Hat das Kind ein Deletions-Syndrom mit Translokation oder Inversion und sind im Karyogramm von Mutter oder Vater entsprechende balancierte Auffälligkeiten zu erkennen, erhöht sich die Wahrscheinlichkeit, dass es bei der Zeugung eines weiteren Kindes erneut zu einer Inbalance des Erbgutes kommt. Die Eltern haben die Möglichkeit, sich vor und während einer Schwangerschaft humangenetisch beraten zu lassen und zur vorgeburtlichen Überprüfung des Vorliegens einer Deletion beim Kind entsprechende Untersuchungsverfahren der Pränataldiagnostik in Anspruch zu nehmen. Bei einem positiven Befund ist es ihnen freigestellt, einen Abbruch der Schwangerschaft aus medizinischer Indikation vornehmen zu lassen.
Ein klinisch unauffälliges Kind, dessen Mutter oder Vater eine balancierte Translokation oder Inversion hat, kann diese geerbt haben und wäre in diesem Falle auch ein potentieller Merkmalsträger.
Literatur
- W. Andler: Endokrinologische Störungen bei Deletionen des Chromosoms 18 (MSchr. Kinderk. 140, 2000, Seite 303-306)
- Kindernetzwerk e.V.: Informationsmappe De Grouchy-Syndrom
- Turleau C: Monosomy 18p. Orphanet J Rare Dis. 2008 Feb 19;3:4. PMID 18284672

Wikimedia Foundation.