- Creutzfeldt-Jakob
-
Klassifikation nach ICD-10 A81.- Atypische Virus-Infektionen des Zentralnervensystems A81.0 Creutzfeldt-Jakob-Krankheit F02.1* Demenz bei Creutzfeldt-Jakob-Krankheit ICD-10 online (WHO-Version 2006) Die Creutzfeldt-Jakob-Krankheit (CJK) (engl. Creutzfeldt Jakob Disease, CJD) ist eine beim Menschen sehr selten auftretende, tödlich verlaufende und durch atypische Eiweiße (siehe Prionen) gekennzeichnete Übertragbare spongiforme Enzephalopathie (TSE). Diese Erkrankung kommt beim Menschen als übertragene, genetische oder sporadische Form vor. Charakteristisch für die Krankheit ist, dass die abnorm gefalteten Prionproteine vor allem im Gehirn den dort normalerweise vorhandenen Vettern mit gesunder Struktur ihre veränderte Struktur aufzwingen und so dort einen verhängnisvollen biochemischen Prozess auslösen, der letztlich zu einer Degeneration des Gehirns führt. Die krankhaft gefalteten Proteine lagern sich in Nervenzellen zusammen und bilden Klumpen. Die Funktion der Nervenzellen wird zunehmend gestört, sodass es bis hin zum programmierten Zelltod kommt. Bei fortschreitender Erkrankung nimmt das befallene Gehirn eine schwammartig durchlöcherte Struktur mit fadenförmigen, proteinhaltigen Ablagerungen an. Im Blut eines erkrankten Menschen sind jedoch nur kleinste Mengen der infektiösen Prionen vorhanden. 1920 veröffentlichte der Neurologe Hans-Gerhard Creutzfeldt erstmals zu dieser Krankheit, kurz vor dem Hamburger Neurologen Alfons Maria Jakob. 1922 wurde die Bezeichnung Creutzfeldt-Jakob-Krankheit eingeführt.
Inhaltsverzeichnis
CJK/CJD
Diese Erkrankung ist insgesamt die häufigste beim Menschen vorkommende Transmissible spongiforme Enzephalopathie (TSE). Diese klassische CJK wird in drei bisher bekannte Formen unterteilt:
Sporadische Prionerkrankung (sCJK)
Die sporadische Form der Creutzfeldt-Jakob-Krankheit ist außer im Vereinigten Königreich die häufigste weltweit beim Menschen auftretende Erkrankungsform.
Erreger
Die auslösenden Faktoren sind wahrscheinlich Prionen.
Häufigkeit
Die Erkrankung kommt weltweit mit einer ähnlichen Häufigkeit von etwa 1 Fall pro Jahr pro Million Einwohner vor. In Deutschland beträgt das Geschlechtsverhältnis von Frauen zu Männern 2:1. Das Erkrankungsrisiko nimmt mit steigendem Alter zu und der Erkrankungsgipfel liegt um das 70. Lebensjahr. In diesem Alter beträgt die jährliche Erkrankungswahrscheinlichkeit etwa 1:125.000. Danach sinkt das Risiko wieder ab. Vereinzelt erkranken auch junge Menschen. In Deutschland erkranken jährlich etwa 7 jünger als 50 Jahre alte Menschen an einer sporadischen CJK. Theoretisch gesehen können selbst Jugendliche erkranken, obgleich weltweit bisher nur eine Handvoll derartiger Fälle beschrieben wurde. Die Wahrscheinlichkeit, bereits vor dem 30. Lebensjahr zu erkranken, beträgt etwa 1:3.000.000.
Krankheitsverlauf/Symptome
Die Erkrankung beginnt zunächst schleichend, doch verliert ein Erkrankter unaufhaltsam und rasch fortschreitend seine geistigen und motorischen Fähigkeiten. Folgende Symptome können beobachtet werden: Schreckhaftigkeit, motorische Störungen (Myoklonien, Ataxie), Gedächtnisstörungen, Störungen der Wahrnehmung (Halluzinationen) und der Vigilanz, visuelle Störungen und Persönlichkeitsveränderungen, vegetative Störungen und Verwirrtheit bis hin zur Demenz. Das Spätstadium der Erkrankung ist durch den akinetischen Mutismus gekennzeichnet. In der Regel führt die Erkrankung innerhalb weniger Monate zum Tode. Die Erkrankungsdauer kann von 3–6 Wochen bis zu mehr als 2 Jahren betragen. Die mittlere Erkrankungsdauer beträgt 4–6 Monate. Eine deutliche Häufung wird im 6.–7. Lebensjahrzehnt beobachtet.
Genetische Prionerkrankung
In dieser Form wird eine ganze Gruppe von familiär vererbbaren Erkrankungen zusammengefasst. Bei all diesen Formen wird eine spezifische Mutation vererbt, welche zu einem fehlerhaften Prion-Protein führt. Diese Krankheitsgruppe ist sehr uneinheitlich (heterogen) und sie ist durch sehr variable klinische Symptome gekennzeichnet. Der Erkrankungsgipfel liegt hier insgesamt um das 50. Lebensjahr und damit früher als bei sporadischer Form. Auch die Erkrankungsdauer ist häufig länger. Zu diesen Erkrankungsformen zählen die familiäre/genetische Creutzfeldt-Jakob-Krankheit, das Gerstmann-Sträussler-Scheinker-Syndrom (GSS) und die tödliche familiäre Schlaflosigkeit (fatal familial insomnia, FFI).
Übertragene Formen
Eine direkte Übertragung des Erregers von Mensch zu Mensch ist bisher nur auf iatrogenem Wege (durch Ärzte verursacht) über Kontakt mit infektiösem Gewebe nachgewiesen worden. Dies geschah besonders früher durch Hirnhaut- und Augenhornhauttransplantate, sowie durch unzureichend sterilisierte neurochirurgische Instrumente. Außerdem wurde eine direkte Übertragung bei aus Leichenhypophysen extrahierten Wachstumshormonen bzw. Gonadotropinen beobachtet. Weltweit sind insgesamt 132 Fälle einer Infektion durch Wachstumshormonpräparate bekannt, wobei die meisten dieser Fälle aus Frankreich und Großbritannien berichtet wurden. In Deutschland wurde bislang noch kein Fall bekannt, obwohl auch hierzulande kleinwüchsige Patienten mit Wachstumshormonen behandelt wurden.
Die meisten Fälle, die mit Hirnhauttransplantaten in Verbindung gebracht werden, traten in Japan auf. Diese Erkrankungen werden fast ausschließlich auf das deutsche Produkt Lyodura von der B.Braun Melsungen AG zurückgeführt. Aufgrund mangelnder Kontrollen der Hirnhautspender, sowie des Herstellungsprozesses, bei dem Hirnhäute ungenügend desinfiziert und in Stapeln übereinandergelagert wurden, wodurch es weiterhin zu einer Querkontamination gesunder Hirnhäute mit Prionen kam, galt dieses Produkt als besonders gefährlich. Lyodura wurde als eine Art „Pflaster“ nicht nur zur Rekonstruktion der Hirnhaut, sondern auch in einer Vielzahl nicht-neurologischer Operationen verwendet, zumal es sich durch geringe Abstoßungsreaktionen auszeichnete. Lyodura musste 1996 aus dem Verkehr gezogen werden.
Es gibt Spekulationen über diverse Einzelfälle, bei denen die Krankheit durch ärztliches Verschulden (iatrogen) hätte übertragen worden sein können: Es wurde zum Beispiel von einem Creutzfeldt Jakob-Opfer berichtet, das wenige Jahre vor der Erkrankung eine Kollagen-Plastik am Schädelknochen erhalten hatte. Das Kollagen stammte vom Rind. Ein vCJD-Opfer hatte sechs Jahre vor Ausbruch der Erkrankung Rindersomatropin injiziert bekommen. Und zwei weitere vCJD-Opfer aus Großbritannien hatten eine Polio-Schluckimpfung erhalten, die aus demselben Kälberserum hergestellt worden war. Auch wurde ein anscheinend erhöhtes CJD-Risiko nach Augeninnendruckmessungen mit einem Goldmann-Tonometer festgestellt.
vCJD
Großbritannien gab am 20. März 1996 bekannt, dass mehrere junge Menschen an einer neuen Variante von CJD gestorben waren.
Übertragung
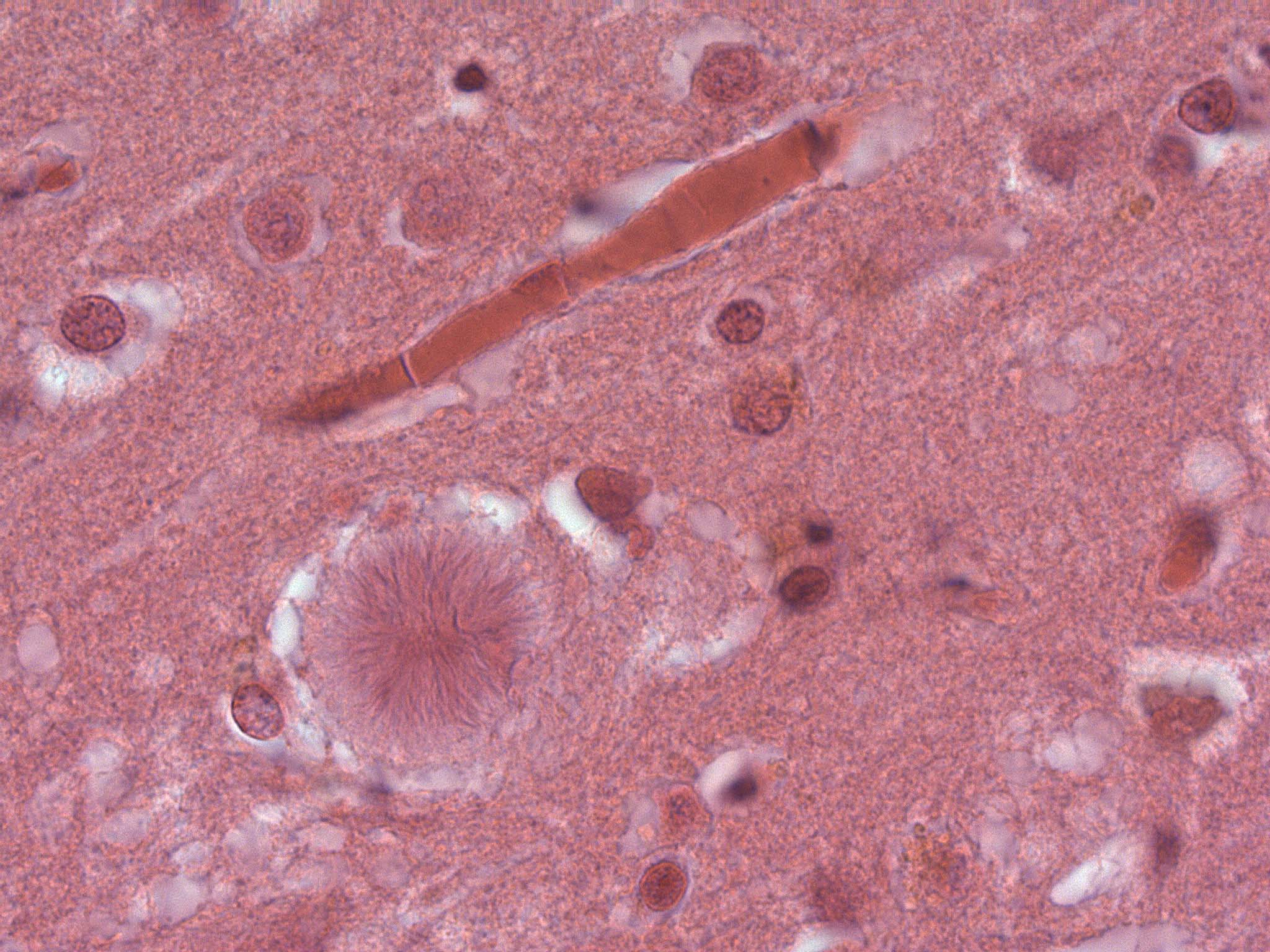
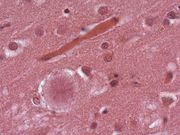 Sog. Florider Plaque in der Hirnrinde (Autopsiepräparat) eines Patienten mit der neuen Variante der Creutzfeldt-Jakob-Krankheit (vCJD)
Sog. Florider Plaque in der Hirnrinde (Autopsiepräparat) eines Patienten mit der neuen Variante der Creutzfeldt-Jakob-Krankheit (vCJD)Nach aktuellen Erkenntnissen besteht eine Wahrscheinlichkeit von 99 % dafür, dass diese Variante (heute als vCJD bekannt) durch den Verzehr von BSE-verseuchtem Rindfleisch hervorgerufen wird. Vermutlich ist allerdings ein Großteil der Bevölkerung gegen die Ansteckung durch BSE-verseuchte Nahrung resistent, denn alle bisherigen vCJD-Erkrankten hatten eine genetische Veranlagung, die sich nur bei knapp 40 Prozent der europäischen Bevölkerung findet. An einer kritischen Stelle des Gens, welches das Prionen-Eiweiß kodiert, fand sich bei ihnen stets nur die Anweisung zum Einbau der Aminosäure Methionin. Der überwiegende Teil der Bevölkerung ist jedoch mischerbig und besitzt zusätzlich ein Gen, das den Einbau von Valin an dieser Stelle bewirkt. Es liegt die Schlussfolgerung nahe, dass sich menschliche Prionen leichter von BSE-Prionen umfalten lassen, wenn sie an der bezeichneten Stelle die Aminosäure Methionin enthalten.
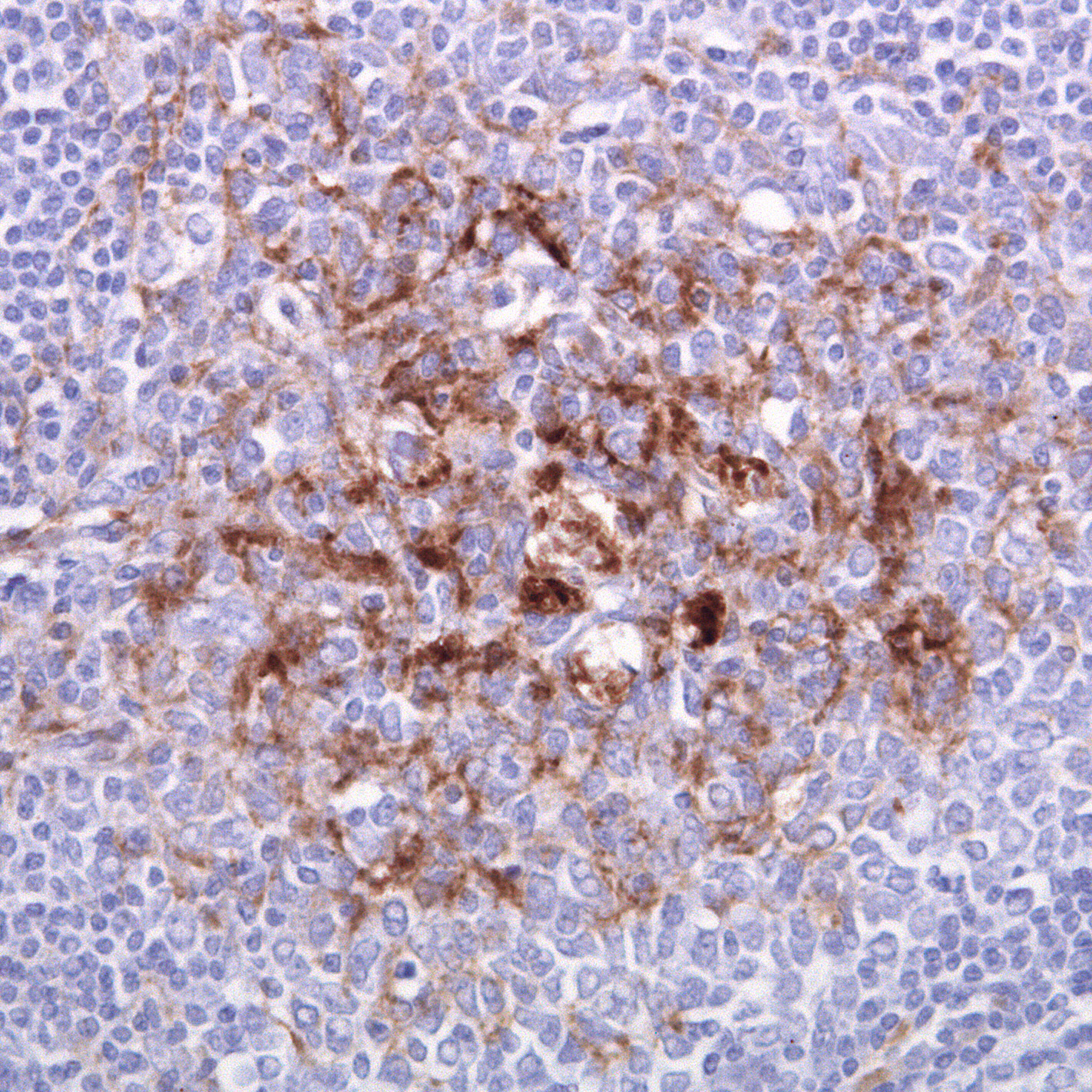
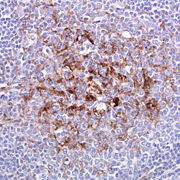 Tonsillenbiopsie eines Patienten mit vCJD. Die braunen Strukturen sind follikulär-dendritische Zellen, in denen das abnormale Prion Protein enthalten ist. (Immunhistochemische Färbung mit Antikörper ICSM35 gegen Prion Protein)
Tonsillenbiopsie eines Patienten mit vCJD. Die braunen Strukturen sind follikulär-dendritische Zellen, in denen das abnormale Prion Protein enthalten ist. (Immunhistochemische Färbung mit Antikörper ICSM35 gegen Prion Protein)Im Unterschied zur sporadischen Erkrankung (sCJD), die vom Gehirn selbst ausgeht und im Wesentlichen auf das Zentralnervensystem beschränkt bleibt (geringe Mengen des abnormen Prionproteins wurden auch in Nerven und Muskeln nachgewiesen), bezieht die variant CJD (vCJD) auch das sogenannte lymphoretikuläre System (Lymphknoten, Milz, Tonsillen (Mandeln)) mit ein. Eine Tonsillenbiopsie (Chirurgische Probenentnahme aus den Rachenmandeln) kann daher zur Diagnose der vCJD verwendet werden. Die Tonsillen sind bei vCJD immer positiv, während sie bei sporadischer CJD (sCJD), bei iatrogener CJD (Mensch zu Mensch-Übertragung durch Dura Transplantate oder Hypophysenextrakt) und bei den erblichen Formen immer negativ sind.
Es gibt außerdem Hinweise darauf, dass Menschen grundsätzlich auch über Bluttransfusionen mit der neuen Variante der Creutzfeldt-Jakob-Krankheit (vCJD) infiziert werden können. In Großbritannien gibt es einige wenige auffällige Einzelfälle in denen es sehr plausibel erscheint, dass die Erkrankung auf diesem Wege übertragen worden ist. Auch Tierversuche deuten auf eine hohe Wahrscheinlichkeit dieser Möglichkeit hin. Mit 100%iger Sicherheit nachgewiesen werden konnte dieser Übertragungsweg allerdings bis jetzt dennoch nicht. Aus der vermutlich gegebenen grundsätzlichen Möglichkeit der Ansteckung über Bluttransfusionen (und damit also Blut bzw. Blutbestandteilen an sich) wurde außerdem die Hypothese abgeleitet, dass es bei einer (allerdings wohl ziemlich unwahrscheinlichen) Verkettung verschiedener unglücklicher Umstände und Zufälle in der Vergangenheit in Großbritannien außerdem bei Operationen jeglicher Art - unabhängig davon ob es zu Bluttransfusionen kam oder nicht - in extrem seltenen Einzelfällen zu einer Infektion durch OP-Besteck gekommen sein könnte. Begründet wird dies damit, dass in der Vergangenheit teilweise Desinfektionstechniken verwendet wurden, bei denen nicht mit Sicherheit ausgeschlossen werden kann, dass Prionen die Prozedur und anschließende Lagerung der Geräte überdauerten. Allerdings ist bisher kein einziger Fall bekannt, in dem ein konkreter Verdacht auf diesen Übertragungsweg vorlag, es handelt sich um eine reine Vermutung hinsichtlich eines denkbaren Übertragungsweges. Ob es jemals einen entsprechenden Fall geben wird ist nicht vorauszusagen. Dennoch wurden aufgrund dieser Hypothese sämtliche Personen die vor dem 31. Dezember 2003 in Großbritannien operiert wurden (die Schwere der OP spielt hierbei keine Rolle) in Deutschland vorsorglich auf unbestimmte Zeit von der Blutspende ausgeschlossen. [1] [2]
Diagnose
Im August 2005 gaben der Neurologe Claudio Soto und seine Kollegen von der University of Texas (USA) bekannt, dass nunmehr die Rinderseuche BSE und die neue Variante der Creutzfeldt-Jakob-Krankheit mit einem Bluttest zu diagnostizieren ist. Die Eigenschaft der abnorm veränderten infektiösen Prionen ihre Struktur anderen gesunden Prionen aufzuzwingen, nutzten die Forscher aus, um die im Blut von Erkrankten nur in verschwindend geringer Zahl vorhandenen infektiösen Prionen um den Faktor zehn Millionen zu vermehren und damit leicht nachweisbar zu machen. In Versuchsreihen mit Hamstern ließen sich so die infektiösen Prionen mit einer Zuverlässigkeit von 89 % und ohne Fehlalarm nachweisen. An der Anwendbarkeit auch für die Diagnose beim Menschen und einer Kontrolle von Blutspenden wird gearbeitet.
Das falsch gefaltete Protein ist gegen die Aufspaltung durch Proteasen resistent, nicht jedoch die normale, gesunde Form von PrP. In der Diagnostik können also die normalen Proteine „verdaut“ werden; und wenn Reste auftreten, dann muss es sich um die pathogene Form des Proteins handeln.
Abgesehen davon lassen sich vCJD-Prionen außerdem im Mandel-, Milz- oder Blinddarmgewebe nachweisen, lange bevor sie das Gehirn befallen. Zur Erhärtung des vCJD-Verdachts führt man daher routinemäßig Mandelbiopsien durch. Schon früh nach Ausbruch der Erkrankung zeigt sich zudem im MRT meist ein bestimmtes Signal in der Pulvinar-Region des Thalamus im Gehirn eines Betroffenen. Bei Vorhandensein dieses Zeichens gilt die Diagnose „vCJD“ als sehr wahrscheinlich.
Krankheitsverlauf/Symptome
vCJD unterscheidet sich von klassischer CJD durch das Alter der Patienten von im Durchschnitt 28 Jahren im Vergleich zu 65 Jahren bei der klassischen CJD, durch andere Symptome und einen längeren Krankheitsverlauf von 14 Monaten im Vergleich zu 6 Monaten bei klassischer CJD. Für den Fall, dass die Krankheit durch den Verzehr von Rindfleisch übertragen wird, rechnen Epidemiologen mit einer durchschnittlichen Inkubationszeit von 12,6 Jahren. Die Erkrankten zeigen meistens zunächst psychische Symptome, wie Depressionen, Wahnvorstellungen, Stimmungsschwankungen oder Angstzustände. Bereits in diesem Anfangsstadium ist das Kurzzeitgedächtnis gestört. Die Patienten klagen über Müdigkeit und die psychischen Symptome verschlechtern sich progressiv und sprechen meist nicht auf medikamentöse Behandlung an. Nach einigen Monaten kommt es in den meisten Fällen zu andauernden schmerzhaften Missempfindungen (Dysästhesien) am ganzen Körper und auch zu Schwindel und Übelkeit. Anschließend treten Koordinationsprobleme (Ataxie) und andere Bewegungsstörungen wie Zittern, Nystagmus, Dystonie, Lähmungen oder unwillkürliche Muskelzuckungen und -bewegungen (Faszikulationen, Myoklonien, Chorea) sowie epileptische Anfälle auf. Außerdem kommt es zu Harn- und Stuhlinkontinenz. Eine Fehlregulation des Muskeltonus bewirkt Gliederschmerzen und es tritt nun auch die für CJD typische, anfangs schleichende, später rasch fortschreitende Demenz in den Vordergrund. In diesem Verlauf kommt es dann zu Halluzinationen und Verwirrtheit, bei denen oft nahe Verwandte nicht mehr erkannt werden. Die Erkrankung verläuft nun akut und führt in nur wenigen Monaten zum vollständigen Zerfall aller Gehirnfunktionen. Die Patienten verweigern die Nahrungsaufnahme und können nur noch durch eine Magensonde ernährt werden. Gelegentlich sterben die Patienten in dieser Phase an vegetativen Störungen oder fallen ins Koma.
Im Endstadium von vCJD haben die Opfer der Krankheit keinerlei Möglichkeit mehr, Kontakt mit ihrer Umwelt aufzunehmen oder auf einen solchen zu reagieren. Darum werden vCJD-Kranke im Endstadium der Krankheit oft als „The Living Dead“ (Die lebenden Toten) bezeichnet. Manchmal tritt hierbei eine vollständige spastische Lähmung des Körpers, die sogenannte Enthirnungsstarre, ein. Die Patienten verweilen recht lange in diesem Endzustand der Erkrankung (terminalen Zustand), bis sie entweder an einer Lungenentzündung oder durch Atemlähmung sterben.
Häufigkeit
Bis zum November 2005 sind in Großbritannien 152 Menschen an vCJD erkrankt, sechs Patienten leben noch. Die Zahl der Erkrankungen nimmt derzeit ab, daher gilt die Gefahr als gebannt. Es wurde jedoch vermutet, dass in den nächsten zehn Jahren eine massive Epidemie des „menschlichen Rinderwahnsinns“ Großbritannien heimsuchen wird und womöglich viele Tausend Menschen dieser Krankheit erliegen werden. Erhärtet wurde diese Vermutung auch durch eine Studie, bei der durch Untersuchungen von entferntem Mandel- und Blinddarmgewebe festgestellt wurde, dass mehrere Tausend Briten den vCJD-Erreger in sich tragen müssen.
Die BBC berichtete allerdings am 12. Januar 2005 von Ergebnissen einer Gruppe von Wissenschaftlern, nach denen eine große Epidemie unwahrscheinlich ist. Dies wird u. a. dadurch gestützt, dass die Zahl der Todesfälle in Großbritannien von 28 im Jahr 2000 auf neun im Jahr 2004 zurückgegangen ist. Ein erneuter Anstieg der Todesfälle kann jedoch nicht ausgeschlossen werden.
Auch in Frankreich sind bereits offiziell 15 Menschen an vCJD erkrankt. Epidemiologen führen die Erkrankungen fast ausschließlich auf importiertes britisches Rindfleisch zurück. Sie erwarten für Frankreich insgesamt etwa 50 Todesfälle. Als größter Risikofaktor gilt hierbei der Verzehr von Fast Food mit Separatorenfleisch-Bestandteil (Hamburger, Döner). Ein Großteil der Betroffenen wird voraussichtlich innerhalb der nächsten zwei Jahre sterben und dürfte bereits jetzt (November 2005) erste Symptome zeigen. Darüber hinaus sind in letzter Zeit in einigen weiteren Europäischen Ländern (Spanien, Italien, Niederlande) erstmals vCJD-Fälle aufgetreten.
Die am meisten gefährdeten Menschen sind zwischen 1980 und 1990 geboren.
Es gibt einen bekannten Fall, in dem ein an vCJD erkrankter Mensch vollständig genesen ist. Es handelt sich um einen amerikanischen Missionar (Don Vanderhoof), dem im Jahr 2000 sowohl in Deutschland als auch in Amerika vCJD diagnostiziert wurde. Medizinisch ist dies eigentlich ausgeschlossen. Am wahrscheinlichsten dürfte es sich hierbei um eine Fehldiagnose handeln.
Einzelnachweise
- ↑ http://www.rki.de/cln_048/nn_196658/SharedDocs/FAQ/NeueVarianteCJK/FAQ__08.html (von dort Links zu ausführlicheren Infos)
- ↑ Wroe SJ, Pal S, Siddique D, HClinical presentation and pre-mortem diagnosis of variant Creutzfeldt-Jakob disease associated with blood transfusion: a case report. Lancet. 2006 Dec 9;368(9552):2061-7:
Weblinks
- Creutzfeldt-Jakob-Krankheit / Variante Creutzfeldt-Jakob-Krankheit – Informationen des Robert Koch-Instituts
- Prionforschungsgruppe Göttingen (Deutsche CJD-Surveillance)
- Leitlinie Creutzfeldt-Jakob-Krankheit der Deutschen Gesellschaft für Neurologie bei AWMF online (Stand 10/2005)
- Uni Düsseldorf: Creutzfeldt-Jakob-Krankheit
- BBC-Bericht (englisch) über die Wahrscheinlichkeit einer größeren Epidemie
- Voraussage der Entwicklung der nvCJD-Fallzahlen in Frankreich (derzeit das am stärksten betroffene Land)

Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.