- Diffusion Ordered Spectroscopy
-
 Ein 300-MHz-NMR-Spektrometer
Ein 300-MHz-NMR-SpektrometerDie Kern(spin)resonanzspektroskopie (NMR-Spektroskopie von engl. nuclear magnetic resonance) ist eine spektroskopische Methode, welche die Untersuchung der elektronischen Umgebung einzelner Atome und der Wechselwirkungen mit seinen Nachbaratomen erlaubt. Diese Information ermöglicht oft die Aufklärung der Struktur und Dynamik von Molekülen und wird besonders in der organischen Chemie und in der Biochemie eingesetzt. Leider sind nicht alle Isotope des Periodensystems einer NMR-spektroskopischen Untersuchung zugänglich, sondern nur solche, deren Kernspin von Null verschieden ist. Dies sind alle Isotope, die eine ungerade Nukleonen- oder Ordnungszahl besitzen. Eine der Kernspinresonanzspektroskopie verwandte Methode ist die Elektronenspinresonanz(-ESR)-Spektroskopie.
Wissenschaftlicher Hintergrund
Teilchen und Atomkerne, die einen Spin
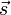 besitzen, verhalten sich im Magnetfeld als hätten sie ein magnetisches Moment, das oft mit
besitzen, verhalten sich im Magnetfeld als hätten sie ein magnetisches Moment, das oft mit 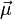 bezeichnet wird. Dieses magnetische Moment kann im äußeren Magnetfeld nicht jede beliebige sondern nur bestimmte, durch die Quantenmechanik bedingte, Orientierungen einnehmen. Die Zahl der möglichen Orientierungen wird durch den Kernspin S bestimmt und beträgt 2S +1 (siehe Multiplizität), das magnetische Moment des Wasserstoffkerns kann beispielsweise aufgrund seines Kernspins von ½ die beiden Zustände +½ und -½ einnehmen, das Deuteriumatom hat dagegen einen Kernspin von 1, hier sind die Zustände +1, 0 und -1 möglich. Die (sehr kleine) Energiedifferenz zwischen zwei benachbarten Zuständen,
bezeichnet wird. Dieses magnetische Moment kann im äußeren Magnetfeld nicht jede beliebige sondern nur bestimmte, durch die Quantenmechanik bedingte, Orientierungen einnehmen. Die Zahl der möglichen Orientierungen wird durch den Kernspin S bestimmt und beträgt 2S +1 (siehe Multiplizität), das magnetische Moment des Wasserstoffkerns kann beispielsweise aufgrund seines Kernspins von ½ die beiden Zustände +½ und -½ einnehmen, das Deuteriumatom hat dagegen einen Kernspin von 1, hier sind die Zustände +1, 0 und -1 möglich. Die (sehr kleine) Energiedifferenz zwischen zwei benachbarten Zuständen, 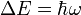 mit der Larmorfrequenz ω, ist proportional zur Stärke des äußeren Magnetfeldes. Aufgrund der Energiedifferenz werden die energieärmeren Zustände bevorzugt besetzt, allerdings ist diese Bevorzugung wegen der nur kleinen Energiedifferenz nur gering, daher braucht man für die NMR-Spektroskopie sehr starke Magnetfelder. Lässt man auf den Kern zusätzlich ein elektromagnetisches Wechselfeld (z.B. Radiowellen), dessen magnetischer Vektor senkrecht auf dem statischen magnetischen Feld steht, einwirken, so kann man einen Übergang in einen höherenergetischen Zustand erzwingen, wenn die eingestrahlte Frequenz der Lamorfrequenz entspricht. Wenn das System gesättigt ist, sind alle Zustände gleich besetzt, alle Kerne in den energiereichsten Zustand zu überführen ist nicht möglich (Boltzmann-Statistik). Zur Messung bringt man die Probe in ein magnetisches Feld. Die Probe wird von einer Induktionsspule umgeben, welche ein hochfrequentes elektromagnetisches Wechselfeld erzeugt. Dann variiert man die Stärke des Magnetfeldes, bis der Resonanzfall eintritt (Continuous-Wave-Verfahren). Alternativ kann auch die magnetische Feldstärke konstant gehalten und die Frequenz des eingestrahlten Wechselfeldes variiert werden (Continuous-Field). Wenn der Resonanzfall eintritt, die Probe also Energie aus dem Wechselfeld aufnimmt, verändert sich die Stromstärke, welche zum Aufbau des Wechselfeldes benötigt wird. Dies kann man messen. Alternativ kann auch die während der Relaxation nach Abschalten des Wechselfeldes abgegebene Strahlung registriert werden (z. B. einem Oszilloskop). Die Energieabstrahlung beim Zurückfallen vom höheren in den energieärmeren Zustand verläuft innerhalb einer sehr kurzen Periode, der sogenannten Relaxationszeit. Diese beträgt bei Raumtemperatur bei Protonen etwa eine halbe Sekunde. Mit einem ständig ein- und ausschaltenden Relais können Radiowellensender und Detektor jedoch kontinuierlich betrieben werden.
mit der Larmorfrequenz ω, ist proportional zur Stärke des äußeren Magnetfeldes. Aufgrund der Energiedifferenz werden die energieärmeren Zustände bevorzugt besetzt, allerdings ist diese Bevorzugung wegen der nur kleinen Energiedifferenz nur gering, daher braucht man für die NMR-Spektroskopie sehr starke Magnetfelder. Lässt man auf den Kern zusätzlich ein elektromagnetisches Wechselfeld (z.B. Radiowellen), dessen magnetischer Vektor senkrecht auf dem statischen magnetischen Feld steht, einwirken, so kann man einen Übergang in einen höherenergetischen Zustand erzwingen, wenn die eingestrahlte Frequenz der Lamorfrequenz entspricht. Wenn das System gesättigt ist, sind alle Zustände gleich besetzt, alle Kerne in den energiereichsten Zustand zu überführen ist nicht möglich (Boltzmann-Statistik). Zur Messung bringt man die Probe in ein magnetisches Feld. Die Probe wird von einer Induktionsspule umgeben, welche ein hochfrequentes elektromagnetisches Wechselfeld erzeugt. Dann variiert man die Stärke des Magnetfeldes, bis der Resonanzfall eintritt (Continuous-Wave-Verfahren). Alternativ kann auch die magnetische Feldstärke konstant gehalten und die Frequenz des eingestrahlten Wechselfeldes variiert werden (Continuous-Field). Wenn der Resonanzfall eintritt, die Probe also Energie aus dem Wechselfeld aufnimmt, verändert sich die Stromstärke, welche zum Aufbau des Wechselfeldes benötigt wird. Dies kann man messen. Alternativ kann auch die während der Relaxation nach Abschalten des Wechselfeldes abgegebene Strahlung registriert werden (z. B. einem Oszilloskop). Die Energieabstrahlung beim Zurückfallen vom höheren in den energieärmeren Zustand verläuft innerhalb einer sehr kurzen Periode, der sogenannten Relaxationszeit. Diese beträgt bei Raumtemperatur bei Protonen etwa eine halbe Sekunde. Mit einem ständig ein- und ausschaltenden Relais können Radiowellensender und Detektor jedoch kontinuierlich betrieben werden.Wenn alle 1H- oder 13C-Atome die gleiche Resonanzfrequenz hätten, wäre die Methode zur Strukturaufklärung wenig interessant. Tatsächlich hängen aber die Resonanzfrequenzen von der elektronischen Umgebung der Kerne ab. Das äußere Magnetfeld induziert in den Elektronenhüllen einen Ringstrom, welcher wiederum ein Magnetfeld erzeugt, das dem äußeren entgegen gerichtet ist und so die Stärke des Magnetfeldes am untersuchten Kern verringert (Abschirmung). Außerdem gibt es kleine Resonanz-Wechselwirkungen über Bindungen mit anderen magnetischen Kernen („chemische Kopplung“). Aufgrund dieser Eigenschaften wird die Kernresonanzspektroskopie zur Strukturaufklärung von Molekülen eingesetzt.
Anwendungsgebiete
NMR-Spektren können am einfachsten für Moleküle aufgenommen werden, die sich in Lösung befinden und nicht mit paramagnetischen Substanzen in Wechselwirkung stehen. NMR-Spektroskopie an paramagnetischen Substanzen und an Festkörpern ist ebenfalls möglich, die Interpretation der Spektren und die Aufbereitung der Proben für die Messung sind aber in beiden Fällen deutlich komplexer. Bezüglich der NMR an Festkörpern vgl. auch Magic Angle Spinning.
Die hochauflösende Kernresonanzspektroskopie in Lösung wird heute in großem Maßstab für folgende Aufgaben verwendet:
- Zum zerstörungsfreien Nachweis von Inhaltsstoffen einer Probe
- Zur Bestimmung von Molekülstrukturen (von kleinen Molekülen bis hin zu Proteinen und Nukleinsäurefragmenten)
- Zur Untersuchung von Wechselwirkungen zwischen Molekülen
Neben spektroskopischen Untersuchungen vermittelt auch die Bestimmung von Kernspin-Relaxationszeiten Informationen über die Struktur und Dynamik von Materialien.
Unterschiedliche Kernspin-Relaxationszeiten in verschiedenen biologischen Geweben bilden auch die Basis für die in der Medizin als bildgebendes diagnostisches Verfahren genutzte Magnetresonanztomographie (Kernspintomographie). Magnetresonanztomographie-Methoden finden außer in der medizinischen Diagnostik auch zunehmend Anwendungen in den Ingenieur- und Geowissenschaften.
Methoden zur Strukturaufklärung in der organischen Chemie
Chemische Verschiebung und Integration von Signalen
Für eine bessere Vergleichbarkeit der Resonanzfrequenzen bezieht man diese auf eine Standardsubstanz, als Standard für 1H- und 13C-NMR-Spektren hat sich heute Tetramethylsilan (TMS) durchgesetzt. Die Methylgruppen des Tetramethylsilans haben nach der Definition eine chemische Verschiebung von Null. Während früher jeder Probe einige Milligramm TMS zugesetzt wurden, bezieht man sich heutzutage in der Regel auf Restprotonen des deuterierten Lösungsmittels.
Die chemische Verschiebung ist definiert als: δ (ppm) = (ν H(gemessen) - νH (TMS)) /Betriebsfrequenz in MHz.
Die chemische Verschiebungen von Wasserstoffkernen in organischen Molekülen wird durch die Art der funktionellen Gruppen beeinflusst. Je nach Art und Besonderheiten des Moleküls weichen die chemischen Verschiebungen trotz gleicher funktioneller Gruppen mitunter ganz leicht von einander ab, so dass die Anordnung aller chemischen Verschiebungen im NMR-Spektrum ganz charakteristisch für eine Substanz ist.
Bei jedem Signal, das durch eine Messkurve dargestellt wird und von einer bestimmten chemischen Verschiebung einer Wasserstoffgruppe stammt, kann auch die Anzahl der daran beteiligten Wasserstoffatome bestimmt werden; sie entspricht dem Flächeninhalt unter der Messkurve. Dieser wird durch Integration errechnet und von einem Signalschreiber am NMR-Spektrometer dargestellt.
Durch Auswertung dieses Integrals kann also beispielsweise bestimmt werden, wie viele Wasserstoffatome eines Moleküls sich an Methylgruppen, an Aromaten, an Carboxylgruppen, an Doppelbindungen usw. befinden. Diese Kenntnis ist für organische Chemiker bei der Bestimmung von Strukturen äußerst wichtig.
Wasserstoffatome Chemische Verschiebung (δ) H3C- 0,9 H3CCR=C 1,6 H3C-Ar 2,3 H3C-CO-R 2,2 H3C-OR 3,3 R2C-CH2-CR2 1,4 -C-CH2-Cl 3,6 R-CO2H 9 bis 13 ROH 0,5 bis 4,5 H-Ar 7,2 Chemische Kopplung
Die chemische Kopplung basiert auf dem Prinzip, dass sich die Wasserstoffkerne oder Kohlenstoffkerne über die chemischen Bindungen untereinander „wahrnehmen“ und daher koppeln können. Die Kopplung reicht jedoch nur zum nächsten Nachbarkohlenstoffatom, der übernächste Nachbar wird in der Regel nicht mehr wahrgenommen und koppelt daher nicht. Auch Wasserstoffatome am selben Kohlenstoffatom zeigen meist keine Kopplung.
Ähnlich wie in einem Wohnblock, in dem man die nahe benachbarten Mieter noch kennt aber über entfernter lebende Bewohner keinerlei Ahnung hat, verhält es sich bei der chemischen Kopplung in der NMR-Resonanzspektroskopie. Da jedoch alle Kopplungen miteinander verbunden sind, d. h. jedes Kohlenstoffatom auch wieder Nachbarn hat, kann ein Chemiker aus einem NMR-Spektrum die gesamten Nachbarschafts- und Strukturverhältnisse eines organischen Moleküls durch die Kopplung erschließen.
- Ein Beispiel:
Nimmt man als einfaches Beispiel das 1H-NMR-Spektrum von 1-Chlorethanol und Ethanol, so koppelt im Falle des 1-Chlorethanol die Methylgruppe mit dem einzelnen Wasserstoffkern, der sich an dem Kohlenstoffatom mit der Hydroxylgruppe und dem Chlor befindet. Der Spin jedes einzelnen Wasserstoffkerns kann sich im energiereichen oder im energieärmeren Zustand befinden. Die beiden unterschiedlichen Magnetfelder, die der einzelne Wasserstoffkern erzeugt, führen zu einer Aufspaltung in zwei gleich große Signale bei der Methylgruppe. Man nennt die Aufspaltung in zwei gleichgroße Signale ein Dublett, der Abstand zwischen den beiden Signalen heißt Kopplungskonstante (in diesem Falle ca. 7 Hz).
Beim Ethanol „sieht“ die Methylgruppe zwei Wasserstoffkerne der Methylengruppe. Es entstehen zwei Dubletts. Wenn die Lage der rechten Seite des linken Dubletts genau mit der linken Seite des rechten Dubletts übereinstimmt, entsteht ein Triplett - also drei Signalpeaks - mit dem Flächenverhältnis von 1:2:1 und einer Kopplungskonstante von 7 Hz.
Die Methylengruppe im Ethanol „sieht“ die benachbarten 3 Wasserstoffkerne der Methylgruppe als Quartett - es zeigen sich vier Signalpeaks - mit einer Aufspaltung im Verhältnis von 1:3:3:1. Der Wasserstoffkern der Hydroxylgruppe koppelt nicht, er erscheint als Singulett.
Für den Erhalt von Strukturinformationen bei sehr komplexen Molekülen kann auch ein Signal „entkoppelt“ werden, d. h. man hebt durch Einstrahlung einer bestimmten Frequenz die Kopplung zu seinen Nachbarn auf, dadurch verändert sich das Kopplungsmuster der Kernspins der näheren Umgebung.
Funktionsprinzip
Technisches zur NMR-Spektroskopie
Früher wurden NMR-Resonanzen mit einer Brückenschaltung in Schwingkreisen bestimmt. Bloch und Mitarbeiter nutzten zwei identische Schwingkreise, d. h. zwei Spulen und zwei Kondensatoren, um einen Abgleich mit einer Brückenschaltung vorzunehmen; eine Spule als Sender eine als Empfänger. Es ist aber auch möglich, eine Brückenschaltung mit nur einer Spule herzustellen. Dieses Verfahren wurde von Purcell genutzt.
Vor der Probenvermessung wird die Brücke mit der zu messenden Frequenz abgeglichen. Mit Gleichungen aus der Physik kann man für einen Schwingkreis und eine Brückenschaltung die Phasenverschiebung zwischen Strom und Spannung, den Scheinwiderstand und die Stromlosigkeitsbedingungen einer Brücke berechnen.
In die Spule kommt nun ein Substanzröhrchen hinein. Ein Magnetfeld (mit einem Permanentmagneten oder Elektromagneten) wird dann horizontal zur Spulenachse erzeugt. Bei einer ganz bestimmten Frequenz und einer bestimmten Magnetfeldstärke und nur bei Anwesenheit einer Substanzprobe (mit entsprechenden Atomkernen) wird der Schwingkreis verstimmt. Im Oszilloskop oder mit einem Schreiber ist diese Verstimmung sichtbar.
Wasserstoffkerne konnten auch bei sehr geringen Frequenzen (wenige kHz) und einem sehr schwachen Magnetfeld durch Schwingkreisverstimmung nachgewiesen werden. Interessant wird die Methode für die Strukturaufklärung von komplexen Molekülen jedoch erst bei hohen Frequenzen (ab 60 MHz) und stärkeren Magnetfeldern (1,4 Tesla), da sich dann die chemischen Verschiebungen von unterschiedlichen Wasserstoffatomen komplizierten Verbindungen deutlicher unterscheiden. Will man jedoch nicht nur ein einziges Signal auf dem Oszilloskop sehen, sondern mehrere unterschiedliche Wasserstoffatomkerne (oder andere Kerne) so muss ein ganzes Frequenzband eingestrahlt werden (Wobbeln).
Früher - bis in die siebziger Jahre des letzten Jahrhunderts - nutzten die NMR-Spektrometer das Continuous-Wave-Verfahren (CW), um das Spektrum einer komplexen Verbindung abzutasten.
Heute ist die Puls-Fourier-Transformation (PFT) üblich. Hierbei wird ein Hochfrequenzimpuls eingestrahlt. Dieser Impuls enthält ein ganzes Band an Schwingungen.
Die bereits angesprochene Abhängigkeit der Energieniveaus der Kernspins von der Molekülstruktur rührt in erster Linie von der Wechselwirkung der Elektronenstruktur der Moleküle mit dem äußeren Magnetfeld her: Hierdurch entsteht in der Elektronenhülle ein Induktionsstrom, welcher wiederum ein Magnetfeld erzeugt, das dem äußeren entgegen gerichtet ist. Dadurch wird das Magnetfeld am Atomkern geschwächt, die Frequenz der für den Übergang notwendigen Strahlung ist also kleiner als im Falle eines nackten Atomkerns. Die Differenz heißt chemische Verschiebung und wird üblicherweise im Verhältnis zur für den nackten Atomkern nötigen Frequenz angegeben. Chemische Verschiebungen liegen üblicherweise im Bereich von 0–5000 ppm.
Das magnetische Feld wird am Atomkern durch die Ausrichtung weiterer magnetischer Momente in der unmittelbaren Umgebung beeinflusst. Befindet sich beispielsweise ein Kern mit zwei Ausrichtungsmöglichkeiten in der Nähe, so kann dieser das Feld verstärken oder abschwächen. Dies führt zu einer Aufspaltung des Signals, man spricht von einer Kopplung. Weil die chemische Verschiebung im wesentlichen von der Elektronendichte am Atomkern abhängt, kann man für Atomkerne in chemisch ähnlichen Umgebungen auch ähnliche Verschiebungen erwarten. Aus der Kopplung erhält man zusätzlich Informationen über Nachbarschaftsbeziehungen zwischen verschiedenen Kernen in einem Molekül. Beides zusammengenommen liefert wesentliche Hinweise über die Struktur des gesamten Moleküls.
Atomkerne mit einer ungeraden Protonen- und/oder Neutronen-Zahl besitzen einen Kernspin I. Dieser kann ganz- und halbzahlige Werte (z. B. 1/2, 1, 3/2,…9/2) annehmen, bei Isotopen mit gerader Protonen- und Neutronenzahl (sogenannten gg-Kernen) ist er 0. Von Null verschiedene Kernspins gehen mit einem magnetischen Dipolmoment einher. Die Größe dieses Dipolmoments wird durch das gyromagnetische Verhältnis des betreffenden Isotops beschrieben. In einem äußeren, statischen Magnetfeld richten sich magnetische Kernmomente entsprechend den Regeln der Quantenmechanik aus. Ein Atomkern mit I = 1/2 hat die Form einer Kugel, Kerne mit I > 1/2 haben eine ellipsoidische Form und haben daher zusätzlich ein elektrisches Quadrupolmoment „eQ“, welches mit elektrischen Feldgradienten wechselwirken kann (siehe auch NQR). Diese zusätzliche starke, elektrische Wechselwirkungsmöglichkeit führt zu breiten NMR-Resonanzlinien, die komplizierter zu interpretieren sind als die schmalen, durch gut auflösbare Kopplungen strukturierten Resonanzlinien der Spin-1/2-Kerne.
Die am meisten für die chemische Strukturaufklärung genutzten Isotope sind daher Kerne mit Spin 1/2. Hierzu gehören unter anderem die Nuklide 1H, 13C, 15N, 19F, 29Si und 31P. Spin-1/2 Kerne können nur zwei diskrete Zustände annehmen, nämlich entweder parallel oder antiparallel zum äußeren Magnetfeld. Zwischenstellungen sind quantenmechanisch verboten. Die zwei Anordnungsmöglichkeiten entsprechen zwei unterschiedlichen Energiezuständen.
Die Energiedifferenz zwischen diesen beiden Zuständen ist proportional zur Stärke des Magnetfelds am Kernort. Der Proportionalitätsfaktor ist dabei das gyromagnetische Verhältnis des betreffenden Isotops. Übergänge zwischen den beiden Orientierungen der Kernmomente können durch die Einstrahlung resonanter magnetischer Wechselfelder ausgelöst werden. Die Resonanzfrequenz ist der Energieaufspaltung zwischen den beiden Kernspins proportional und wird auch als Larmorfrequenz bezeichnet.
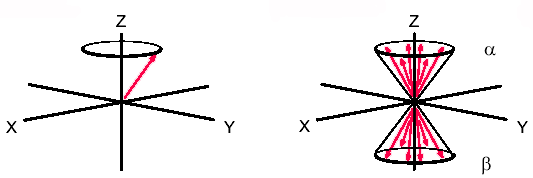
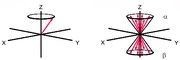 Spindiagramm eines Atoms und mehrerer Atome
Spindiagramm eines Atoms und mehrerer AtomeVeranschaulichen lässt sich dies durch das nebenstehende Diagramm. Hierbei denkt man sich ein Koordinatensystem mit dem äußeren Magnetfeld entlang der z-Achse. Ein Atomkern mit einem Spin von 1/2 richtet sich mit einem Spin-Vektor entweder parallel oder antiparallel zum äußeren Feld aus. Wenn man nun die Vektoren mehrerer Atome in dieses Koordinatensystem aufnimmt, entstehen zwei Kegel, jeweils einer für parallel und antiparallel. Infolge des Energie-Unterschieds zwischen der parallelen und der antiparallelen Orientierung der magnetischen Kernmomente gibt es im thermischen Gleichgewicht einen Besetzungsunterschied zwischen den beiden Orientierungen. Dieser folgt in Hochtemperatur-Näherung der Boltzmann-Verteilung und bewirkt eine Überschussmagnetisierung in positiver Richtung entlang der z-Achse.
Das NMR-Signal kommt dadurch zustande, dass man die zu untersuchende Probe im Magnetfeld einem Radiofrequenz-Puls aussetzt. Dabei werden die Spins der einzelnen Atome durch das Magnetfeld des Pulses beeinflusst, so dass der Gesamtvektor, der sich aus den gezeigten Spin-Kegeln ergibt, gekippt wird. Er liegt nun nicht mehr parallel zur z-Achse, sondern ist um einen Winkel ausgelenkt, der proportional zur Dauer und Intensität der Radiofrequenzpulses ist. Typisch sind Pulslängen von etwa 1–10 µs. Eine maximale Quermagnetisierung senkrecht zur z-Achse wird bei einem Auslenkungswinkel von 90° erreicht.
Diese Quermagnetisierung verhält sich wie ein magnetischer Kreisel und präzediert in der Ebene senkrecht zum statischen Magnetfeld. Diese Präzessionsbewegung macht sich als sehr schwaches magnetisches Wechselfeld bemerkbar, das mittels geeigneter Verstärker gemessen wird. Nach Beenden der resonanten Einstrahlung treten zwei Prozesse, sogenannte Relaxationsprozesse, ein, durch die die Quermagnetisierung wieder abnimmt. Das NMR-Signal wird also nach Beenden des Radiofrequenzpulses als Freier Induktionszerfall (FID; von englisch: free induction decay) gemessen. Die Zeitkonstante T2* dieses freien Induktionszerfalls hängt von der transversalen Relaxationszeit T2 sowie von der Homogenität des Magnetfelds ab. Für leicht bewegliche Flüssigkeiten in homogenen Magnetfeldern kann sie im Bereich von mehreren Sekunden liegen. Der FID wird durch die Frequenzunterschiede infolge von chemischer Verschiebung und Kopplung moduliert. Durch eine Fourier-Transformation kann die Verteilung der verschiedenen Frequenzen aus dem FID berechnet werden. Dies ist dann das NMR-Spektrum. Das NMR-Spektrum liefert in vielen Fällen einen eindeutigen „Fingerabdruck“ des jeweiligen Moleküls. Zusammen mit Informationen aus weiteren Messungen wie z. B. der Massenspektrometrie kann aus den Spektren Strukturaufklärung die chemische Struktur einer unbekannten Substanz bestimmt werden.
Kommerzielle NMR-Spektrometer für die chemische Strukturaufklärung arbeiten üblicherweise bei Feldstärken zwischen 7 und 21 Tesla. Für 1H entsprechen die Resonanzfrequenzen (Larmorfrequenzen) dann zwischen 300 und 900 MHz. Da 1H der wichtigste NMR-Kern ist, wird die Feldstärke von Spektrometern gewöhnlich in dessen Larmorfrequenz ausgedrückt. Bei 1H beträgt die Aufspaltung der Spektren infolge unterschiedlicher chemischer Verschiebungen ca. 10 ppm. Dies entspricht also einer maximalen Bandbreite von ca. 3 kHz bei einer NMR-Frequenz von 300 MHz. Die Frequenzbandbreite der NMR-Spektren infolge der chemischen Verschiebung wächst proportional zum Magnetfeld an. Die chemische Verschiebung selbst ist als Verhältnis der Differenz der Resonanzfrequenz des Kerns in einer bestimmten chemischen Umgebung und der Resonanzfrequenz in einer Referenzverbindung zur Resonanzfrequenz selbst definiert. Dies erlaubt einen einfachen Vergleich zwischen NMR-Spektren, die bei verschiedenen Feldern gemessen wurden. Für Wasserstoff und Kohlenstoff wird Tetramethylsilan (TMS)) als Referenzsubstanz genommen. Der Bereich von chemischen Verschiebungen für Kohlenstoff und viele andere Kerne ist wesentlich breiter als für Wasserstoff und kann mehrere 100 ppm betragen. Bei einigen sehr schweren Kernen wie z. B. 207Pb werden auch chemische Verschiebungen im Bereich von % beobachtet.
Empfindlichkeit
Ein inhärentes Problem der NMR-Spektroskopie ist ihre vergleichsweise geringe Empfindlichkeit (schlechtes Signal-Rausch-Verhältnis). Für Messungen sind je nach Experiment und Messzeit ca. 10 nmol bis 1 µmol Substanz notwendig (typische Probenmenge: 1 mL einer Lösung mit einer Konzentration von 10 µmol/L bis 1 mmol/L).
Ursache dafür sind die durch die Boltzmannverteilung festgelegten geringen Besetzungsunterschiede der Energieniveaus:
Mit dieser Gleichung wird das Besetzungsverhältnis
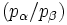 der beiden beteiligten Energiezustände durch deren Energiedifferenz im Verhältnis zur thermischen Energie bei gegebener Temperatur T ausgedrückt. Darin ist k die Boltzmann-Konstante. Die Energiedifferenz entspricht dabei der Energie eines Lichtquants (
der beiden beteiligten Energiezustände durch deren Energiedifferenz im Verhältnis zur thermischen Energie bei gegebener Temperatur T ausgedrückt. Darin ist k die Boltzmann-Konstante. Die Energiedifferenz entspricht dabei der Energie eines Lichtquants (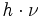 ), das ein Teilchen vom günstigeren in den ungünstigeren Zustand befördert (Grundgleichung der Spektroskopie). Bei einer Resonanzfrequenz von 600 MHz und einer Temperatur von 0 °C bzw. 273 K ergibt sich ein Wert von ungefähr e0,0001, also in etwa gleich eins. Daher sind schon im thermischen Gleichgewicht fast gleich viele Kerne im angeregten Zustand wie im Grundzustand – allein die Wärme sorgt dafür. Zum Vergleich: Sichtbares Licht besitzt um einen Faktor von etwa 1 Million höhere Frequenzen. Folglich haben Übergänge, die durch sichtbares Licht angeregt werden, Besetzungsunterschiede von etwa e100. Das bedeutet, dass praktisch alle Teilchen im Grundzustand sind, was die Spektroskopie im sichtbaren Bereich wesentlich empfindlicher macht.
), das ein Teilchen vom günstigeren in den ungünstigeren Zustand befördert (Grundgleichung der Spektroskopie). Bei einer Resonanzfrequenz von 600 MHz und einer Temperatur von 0 °C bzw. 273 K ergibt sich ein Wert von ungefähr e0,0001, also in etwa gleich eins. Daher sind schon im thermischen Gleichgewicht fast gleich viele Kerne im angeregten Zustand wie im Grundzustand – allein die Wärme sorgt dafür. Zum Vergleich: Sichtbares Licht besitzt um einen Faktor von etwa 1 Million höhere Frequenzen. Folglich haben Übergänge, die durch sichtbares Licht angeregt werden, Besetzungsunterschiede von etwa e100. Das bedeutet, dass praktisch alle Teilchen im Grundzustand sind, was die Spektroskopie im sichtbaren Bereich wesentlich empfindlicher macht.Um die Empfindlichkeit zu steigern, werden verschiedene Wege eingeschlagen:
- Messung möglichst empfindlicher Kernsorten (besonders 1H)
- Mehrfache Messung einer Probe und Addition aller Spektren
- Einsatz stärkerer Magneten (supraleitende Magnete).
- Elektronisches Rauschen durch Kühlung der Empfänger verringern (Cryoelektronik).
- Anreicherung mit magnetischen Kernen, deren natürliche Häufigkeit gering ist (z. B. 13C bzw. 15N). Das wird z. B. bei Proteinen oft gemacht.
- Verwendung von Hyperpolarisationsmethoden, um die Besetzungsunterschiede künstlich zu vergrößern.
Puls-Fourier-Transform NMR
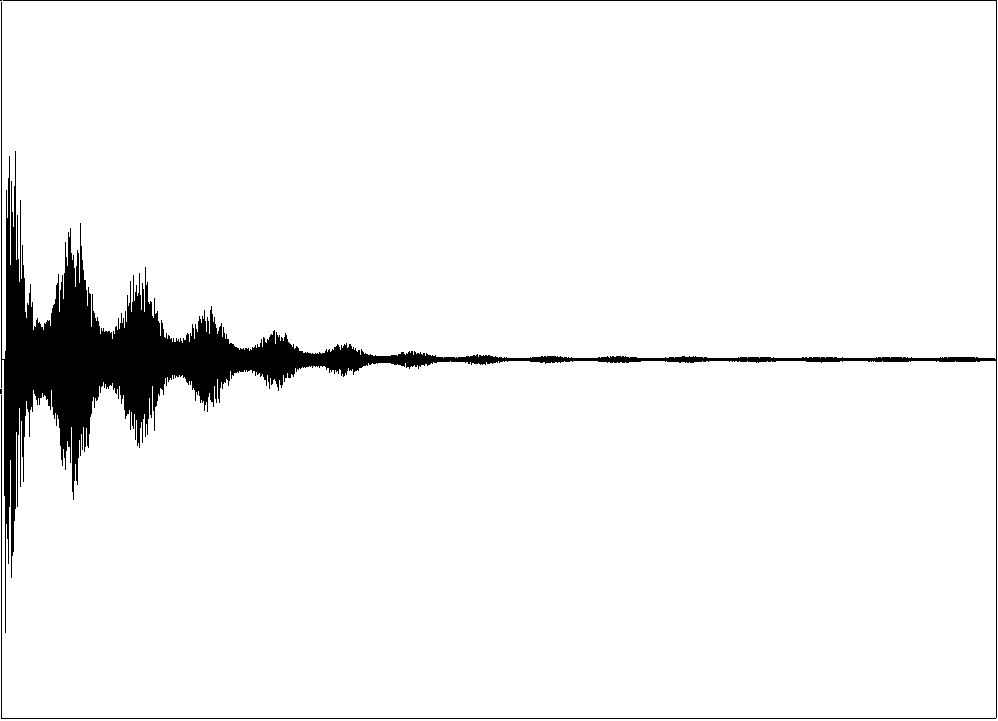
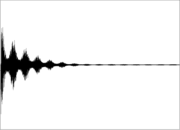 FID
FIDHeutzutage arbeiten alle modernen NMR-Spektrometer mit der Puls-Technik. Bei dieser wird ein einzelner Radiofrequenzimpuls (RF-Puls) oder eine Sequenz von RF-Pulsen auf eine Probe gesandt, die sich in einem starken Magnetfeld befindet. Das FID-Signal nach einer Pulssequenz wird als Funktion der Zeit registriert. Durch Fourier-Transformation wird das Zeitsignal im Computer in das Frequenzspektrum transformiert.
Diese Messtechnik hat das früher verwendete CW-Verfahren (engl. continous wave) fast völlig verdrängt, bei dem eine andauernde Einstrahlung von Radiowellen niedriger Intensität erfolgt, während das Magnetfeld langsam variiert wird (oder wesentlich seltener bei konstantem Magnetfeld die Frequenz der Radiowellen).
Experimentelle Größen
- Die chemische Verschiebung einer Resonanz ist vom lokalen Magnetfeld am Kernort abhängig, das wiederum von der chemischen Umgebung des betrachteten Kerns abhängt.
- Die Intensität einer Resonanz ist proportional zur Konzentration.
- Bei den Relaxationszeiten angeregter Zustände unterscheidet man zwischen longitudinaler Relaxationszeit (Spin-Gitter-Relaxation) und transversaler Relaxationszeit (Spin-Spin Relaxation). Longitudinale Relaxationszeiten bestimmen die Einstellung der Gleichgewichtsmagnetisierung. Die transversalen Relaxationszeiten bestimmen die Linienbreite der Resonanzlinien. Relaxationseffekte geben Aufschluss über vorhandene Wechselwirkungen und molekulare Bewegungen.
- Räumlich benachbarte Kerne wechselwirken miteinander über magnetische Dipol-Dipol-Wechselwirkung (dipolare Kopplung). Diese Wechselwirkung verschwindet in isotropen Lösungen im zeitlichen Mittel.
- Indirekt können Kerne auch über chemische Bindungen miteinander wechselwirken. Diese skalare Kopplung ist für die Aufspaltung der Signale in Multipletts verantwortlich und stellt eine wesentliche Grundlage für die molekulare Strukturbestimmung mit NMR dar. Der Abstand zweier benachbarter Linien eines Multipletts wird als Kopplungskonstante, die in Hertz gemessen wird, bezeichnet.
Eindimensionale NMR-Spektroskopie
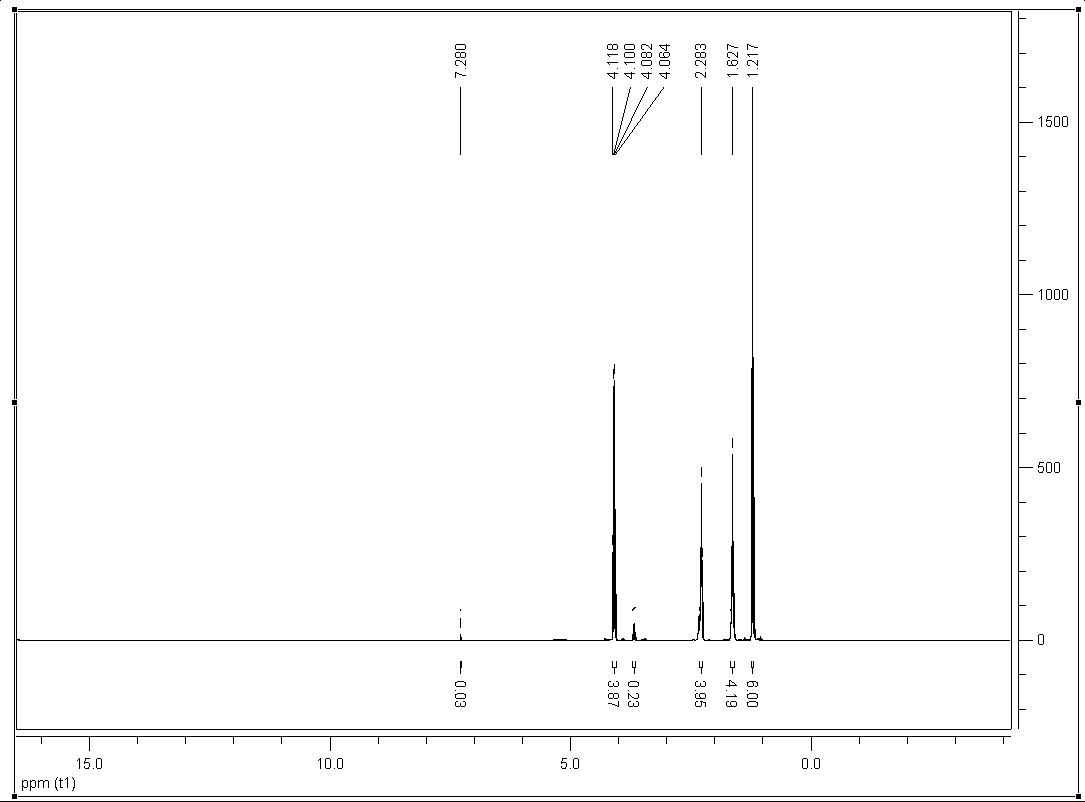
 Typisches NMR-Spektrum
Typisches NMR-SpektrumDie eindimensionale NMR-Spektroskopie ist die am häufigsten angewandte Strukturaufklärungsmethode der Chemie. Bei ihr wird die chemische Verschiebung eines Atoms von einer Referenzsubstanz gemessen. 1H und 13C sind die Kerne, die am häufigsten in der organischen Chemie gemessen werden, aber auch 15N, 31P, 19F und viele andere NMR-aktive Isotope können spektroskopiert werden.
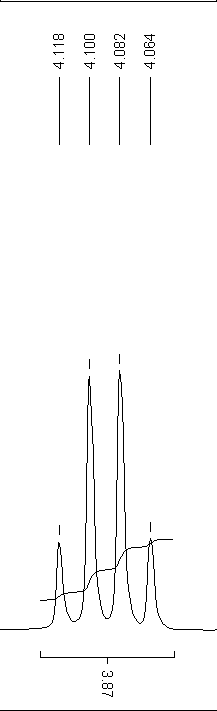
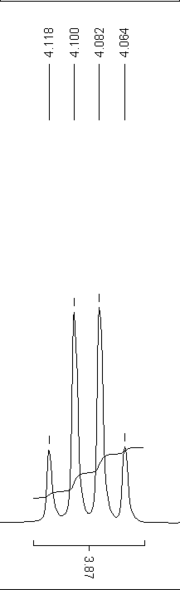 Quartett-Aufspaltung
Quartett-AufspaltungDas Aussehen der Spektren hängt entscheidend von der Aufnahmeart ab. 1H-Spektren werden in der Regel nicht Breitband-entkoppelt aufgenommen. Damit haben alle Wasserstoffatome die Möglichkeit, ihren Spin mit anderen Kernen zu koppeln, die sogenannte Spin-Spin-Kopplung. Damit entsteht bei der charakteristischen chemischen Verschiebung eines Atoms eine für seine Umgebung charakteristische Aufspaltung des Signals, aus der Informationen über die Molekülstruktur abgeleitet werden können.
13C, 15N, 31P, 19F und andere Kerne werden häufig 1H-Breitband-entkoppelt aufgenommen, so dass die Aufspaltung der Signale aufgrund der Kopplungen zu 1H-Kernen ausbleibt.
Spin-Spin-Kopplung
Der Kern eines Atoms kann mit einem benachbarten Atomkern in Wechselwirkung treten. Das kann entweder direkt (durch den Raum) oder indirekt (über die Bindungselektronen zwischen den Kernen) geschehen. Bei einer flüssigen Probe spielt die direkte Kopplung nur eine untergeordnete Rolle, hier treten vor allem Kopplungseffekte über die Bindungen hinweg auf. Die Kopplung entsteht, weil sich die Spins der Bindungselektronen in Nachbarschaft zu einem Kernspin in charakteristischer Weise ausrichten. Der Kern am anderen Ende der Bindung erfährt seinerseits diese Einflussnahme auf die Elektronenspins und nimmt dazu eine antiparallele oder parallele Orientierung ein. Damit gibt es für zwei Kernspins A und B vier Möglichkeiten der Ausrichtung zueinander: (↑↓), (↓↑), (↑↑) und (↓↓).
Abhängig von der Orientierung zum äußeren Magnetfeld wird das effektive Magnetfeld am Kern selber entweder verstärkt (beide Elektronenspins parallel zum Magnetfeld ausgerichtet), abgeschwächt (beide Elektronenspins antiparallel zum Magnetfeld ausgerichtet) oder nicht beeinflusst (ein Spin in, der andere entgegen der Feldrichtung). Da die Kombinationen (↑↓) und (↓↑) energetisch identisch sind, zeigt der Kern Resonanz bei drei Frequenzen. Im Spektrum führt das zu einer Aufspaltung des Signals in drei einzelne Signale. Sind außer dem einen benachbarten Kern noch weitere vorhanden, führt das entsprechend den zusätzlichen Kombinationsmöglichkeiten zu einer weiteren Aufspaltung des Signals. Dabei lässt sich die Anzahl der Signale, die sogenannte Multiplizität, allgemein mit der Formel 2nI+1 berechnen. Darin ist I der Kernspin des betrachteten Kerns und n die Anzahl chemisch äquivalenter koppelnder Kerne. Die relative Intensität der einzelnen Signale kann man dem Pascalschen Dreieck entnehmen.
Sind in obigem Zwei-Kern-Beispiel beide Kerne in einer gleichen chemischen Umgebung, so sind außerdem die Kombinationen (↑↑) und (↓↓) energetisch nicht zu unterscheiden. Man beobachtet nur noch zwei Signale.
Für den Fall, dass ein Kern mit mehreren Kernen unterschiedlicher chemischer Umgebung koppelt, werden die Multiplizitäten, die man für die unterschiedlichen chemischen Umgebungen berechnet, noch einmal miteinander multipliziert. Koppelt beispielsweise ein Kern (I=1/2) mit zwei Kernen die ihrerseits chemisch äquivalent sind und drei weiteren die wiederum untereinander chemisch äquivalent sind, so ergibt sich zunächst für die Kopplung mit den zwei Kernen M=3 und für die drei anderen M=4. Diese Multiplizitäten werden nun multipliziert, womit das Signal des Kerns in zwölf Signale aufspaltet.
Beispiel: Propan und Ethanol
Als ein einfaches Beispiel dient Propan (H3C–CH2–CH3): Die CH2-Gruppe beim Propan hat zwei benachbarte Methylgruppen (–CH3). Dies entspricht sechs benachbarten, äquivalenten H-Atomen. Das Signal wird also in ein Septett aufgespaltet. Bei nichtäquivalenten H-Atomen werden die Nachbaratome einzeln betrachtet, wobei es zu einer Überlagerung der einzelnen Peaks kommen könnte. Um solche Fälle besser auflösen zu können, wird hierfür vielfach auf mehrdimensionale NMR-Techniken wie COSY zurückgegriffen. Der Abstand der einzelnen Peaks in einem Multiplett wird als Kopplungskonstante J bezeichnet. Sie ist unabhängig von dem außen angelegten Magnetfeld.
Die Intensität der einzelnen Signale innerhalb des Multipletts wird durch das Pascalsche Dreieck vorgegeben.
Erklärungen zum Spektrum von Ethanol:
Die OH-Gruppe bildet nur ein Singulett, wenn das Ethanol in wässriger Lösung vorliegt. Das alkoholische Wasserstoffatom ist leicht acid, und kann deswegen ständig durch Wasserstoffatome aus dem Lösungsmittel ausgetauscht werden. Das führt dazu, dass keine permanenten Spin-Spin-Kopplungseffekte auftreten. In reinem Ethanol würde dort, wie wegen der CH2-Gruppe erwartet (M=n+1), ein Triplett entstehen.
Beispiele zweidimensionaler NMR-Spektroskopie
- COSY (engl. correlation spectroscopy)
- Zweidimensionale Methode, bei der gleichartige Kerne (1H) über ihre skalaren Kopplungen miteinander korreliert werden. COSY-Spektren sind symmetrisch bezüglich der Diagonalen. Mit COSY können komplizierte Kopplungsmuster räumlich entzerrt werden.
- DOSY (engl. diffusion ordered spectroscopy)
- Verfahren, bei dem durch das Anlegen von Feldgradienten Moleküle mit unterschiedlichem Diffusionsverhalten NMR-spektroskopisch getrennt erfasst werden können.
- TOCSY (engl. total correlated spectroscopy)
- Zweidimensionale Methode, bei der gleichartige Kerne (1H) über ihre skalaren Kopplungen miteinander korreliert werden. TOCSY-Spektren sind wie COSY-Spektren symmetrisch bezüglich der Diagonalen. Zusätzlich zu den im COSY detektierten Signalen erscheinen im TOCSY auch Korrelationen zwischen dem Startkern und sämtlichen indirekt über mehrere Kopplungen mit ihm verbundenen Kernen (Spinsystem). Das TOCSY-Experiment ist vor allem bei der Strukturaufklärung hochmolekularer Substanzen mit räumlich begrenzten Spinsystemen, wie etwa Polysacchariden oder Peptiden, sehr nützlich.
- HSQC (engl. heteronuclear single quantum coherence)
- Zweidimensionale Methode, bei der chemische Verschiebungen unterschiedlicher miteinander skalar koppelnder Nuklide korreliert werden. Die HSQC-Spektren sind häufig recht übersichtlich, da gewöhnlich nur Signale von direkt aneinander gebundenen Atomen erscheinen. Typische Beispiele sind 1H,13C- und 1H,15N-Korrelationen.
- HMBC (engl. heteronuclear multiple bond coherence)
- Zweidimensionale Methode, bei der chemische Verschiebungen unterschiedlicher miteinander skalar koppelnder Nuklide korreliert werden. Im Gegensatz zum HSQC werden im HMBC Korrelationen über mehrere Bindungen angezeigt. Typisch sind vor allem 1H,13C-Korrelationen.
- NOESY (engl. nuclear overhauser enhancement spectroscopy)
- Zweidimensionale Methode, mit der Korrelationen über den Kern-Overhauser-Effekt (NOE) anstatt über skalare Kopplungen detektiert werden. Mit dieser Methode können räumlich benachbarte Kerne erkannt werden, auch wenn sie nicht skalar miteinander koppeln. Es gibt sowohl homo- als auch heteronukleare Versionen. Dieses Verfahren wird häufig in der Strukturaufklärung eingesetzt.
Historische Entwicklung
Als Ursprung der NMR muss man wohl den experimentellen Nachweis des Protonenspins durch Otto Stern im Jahr 1933 mit einem Molekularstrahlexperiment sehen. Stern hatte bereits vorher zusammen mit Walther Gerlach das berühmte Stern-Gerlach-Experiment entwickelt, mit dem sie 1922 in Frankfurt den schon länger postulierten Elektronenspin nachwiesen. Sie konnten zeigen, dass zunächst ein Strahl von Silberatomen, später dann ein Protonenstrahl, durch ein Magnetfeld in zwei Hälften geteilt wird, die den beiden Spinzuständen zugeschrieben wurden. Stern erhielt für diese Arbeiten den Nobelpreis 1943. Die ersten NMR- und ESR-Experimente führte Isidor Isaac Rabi (Nobelpreis für Physik 1944) mit modifizierten Stern-Gerlach-Anordnungen durch. Er konnte zeigen, dass einer der Halbstrahlen verschwand, wenn man auf ihn mit Hilfe einer Spule ein elektromagnetisches Wechselfeld geeigneter Frequenz (nämlich der Larmorfrequenz) einstrahlte. Das erste erfolgreiche Magnetresonanzexperiment in kondensierter Materie wurde 1944 von E. K. Zavoisky in Kasan an der Wolga durchgeführt. Es handelte sich allerdings nicht um ein NMR-, sondern um ein ESR-Experiment. 1946 veröffentlichten Felix Bloch und Edward Mills Purcell unabhängig voneinander erstmals erfolgreiche NMR-Experimente in flüssiger und fester Phase (Nobelpreis für Physik 1952). Die ersten erfolgreichen NMR-Experimente in Europa wurden 1951 von Harry Pfeifer in Leipzig durchgeführt.
Nachdem kurz darauf die Aufspaltung der Spektren durch chemische Verschiebung und skalare Kopplung erkannt wurde, begann die NMR sich zu einer wichtigen Methode in der chemischen Strukturaufklärung zu entwickeln. Zunächst wurde hauptsächlich die CW-Methode (engl. continuous wave) benutzt, bei der durch Variation der Frequenz oder des Feldes die Resonanzen nacheinander angeregt wurden. 1947 reichten Russell Varian und Felix Bloch ein Patent ein für das erste NMR-Spektrometer. Das erste kommerzielle NMR-Spektrometer wurde dann 1952 von Varian Associates in Palo Alto gebaut. Um 1955 kam die japanische Firma Jeol hinzu und baute ebenfalls NMR-Spektrometer.
Da die CW-Technik allerdings durch ihr schlechtes Signal-Rausch-Verhältnis gekennzeichnet war, entwickelte ab Mitte der 1960er Jahre Richard R. Ernst (Nobelpreis für Chemie 1991) bei der Firma Varian ein Puls-Fourier-Transformation-NMR-Spektrometer (FT-NMR), das eine wesentlich schnellere Aufnahme der Spektren ermöglichte, welche – bei gleicher Messzeit – im Vergleich zu den CW-Spektrometern eine wesentliche Steigerung der Empfindlichkeit (des Signal-Rausch-Verhältnisses) bedeutet. Die ersten kommerziellen NMR-Impulsspektrometer wurden Mitte der 1960er Jahre von der deutschen Firma Bruker (gegründet von Prof. Günther Laukien, einem der NMR Pioniere in Deutschland) in Karlsruhe gebaut. Es folgte die Einführung von Breitbandentkopplung und von Mehrpulsverfahren. Nach einer Idee von Jean Jeener wurden ab Anfang der 1970er Jahre Mehrpulsexperimente mit einer systematisch variierten Wartezeit zwischen zwei Pulsen entwickelt, die nach Fourier-Transformation über zwei Zeitdomänen zu zweidimensionalen Spektren führten. Die Erweiterung zu drei und mehr Dimensionen folgte.
Kurt Wüthrich und viele andere bauten diese 2D- und Multi-Dimensions-NMR zu einer mächtigen Analysetechnik der Biochemie aus, insbesondere zur Strukturanalyse von Biopolymeren wie Proteinen. Wüthrich bekam für diese Arbeiten 2002 den Nobelpreis in Chemie. Im Gegensatz zur Röntgenstrukturanalyse liefert die NMR-Spektroskopie Strukturen von Molekülen in wässriger Lösung. Von besonderer Bedeutung ist die Möglichkeit, detaillierte Informationen über die Moleküldynamik mit Hilfe von Relaxationsparametern zu gewinnen.
Auswertungs-Software
- ACD/Labs Auswertesoftware für 1D- und 2D-NMR sowie zur Vorhersage von NMR-Spektren, mit Datenbankeinbindung
- AUREMOL
- VNMR und VnmrJ (Varian)
- TopSpin (Bruker)
- AMIX (Bruker)
- Mestre-C (Freeware für ältere Releases, Windows) / SwaNMR (MacOS 9, eingestellt) / iNMR (MacOS X)
- NMRPipe (Unix-Systeme)
- SpinWorks
- Delta (JEOL)
- Triad
- NMTNMR (Tecmag)
- PROSPA (Magritek)
- NMRVIEW
- SPARKY – Entwicklung eingestellt
- Nuts (Windows und MacOS 9)
- NPNMR NPNMR 2
- Softwaresammlung, Smartnotebook
- CARA – Freeware (Windows, Linux, Solaris, Irix, OSX), entwickelt in der Gruppe von Prof. Dr. Kurt Wüthrich
- CCPN – The Collaborative Computing Project for NMR
- iNMR ( MacOSX )
- DMFIT – Freeware (Windows, Linux unter Wine), entwickelt in der Gruppe von Prof. Dr. Dominique Massiot
- DAMARIS - entwickelt von Achim Gädke AG Fujara der TU-Darmstadt
Siehe auch
- Elektronenspinresonanz ESR
- Magnetresonanztomographie MRT
- Feldgradienten-NMR
- Medizintechnik
- Apparatemedizin
Weblinks
- Verständliche Animation von BIGS zu Details des MRT, wie Pulssequenzen, Relaxieren oder Spinmodifizierung
- Verständliche Einführung in die Thematik von der TU-Berlin
- www.nmr.de Teil des Webauftritts von www.spectroscopynow.com (John Wiley & Sons)
- Einführung in die 1H-NMR-Spektroskopie
- Spektrendatenbank (nicht nur für NMR)
- Kernspinresonanz – Einführung in die Theorie. Enthält viel Werbung und tote Links.
- Basics of NMR von Joseph P. Hornak, Ph. D.
- e-MRI –Magnetic Resonance Imaging physics and technique course, von Campus Medica
- 1H-NMR Prediction (engl.) - Onlineberechnung von 1H NMR Spectren
- 1H-NMR ohne Formeln - Einführung für den Schulunterricht
- NMR-Parallel-Schwingkreise - Parallelschwingkreise für NMR-Messungen
- James Shoolery, A Basic Guide to NMR, 3rd Edition, Stan's Library, Vol.II, 2008, DOI 10.3247/sl2nmr08.012
Literatur
- Malcom H. Lewitt: Spin Dynamics. 1. Auflage. Wiley&Sons, Chichester 2001, ISBN 978-0471489221.
- Harald Günther: NMR-Spektroskopie. 3. Auflage. Thieme, Stuttgart 1992, ISBN 978-3134875034.
- Ullmanns Enzyklopädie der Technischen Chemie, 4. Auflage, Band 5, S. 382 ff.
- A. Streitwieser, Jr, C.H. Heathcock, Organische Chemie, Verlag Chemie, Basel 1980, S. 205-249
- Dudley H. Williams, Ian Fleming, Spektroskopische Methoden zur Strukturaufklärung, Kernmagnetische Resonanz-Spektren, George Thieme Verlag, Stuttgart 1975, S.80-161

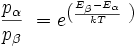
Wikimedia Foundation.