- Epifluoreszenzmikroskop
-
 Fluoreszenzmikroskop
FluoreszenzmikroskopDie Fluoreszenzmikroskopie ist eine spezielle und moderne Form der Lichtmikroskopie.
Inhaltsverzeichnis
Prinzip
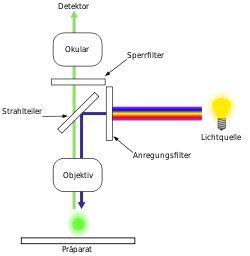 Funktionsschema eines Fluoreszenzmikroskops
Funktionsschema eines FluoreszenzmikroskopsDie Funktion eines Fluoreszenzmikroskopes beruht stets auf folgenden Prinzipien:
- Im zu untersuchenden Präparat befinden sich fluoreszierende Stoffe (Fluorochrome), die mit Licht einer bestimmten Wellenlänge zum Leuchten angeregt werden.
- Die so angeregten Fluorochrome emittieren Licht, welches durch die Stokesverschiebung in der Regel langwelliger als das anregende Licht ist (wichtige Ausnahme: sogenannte 2-Photonenanregung mit einem Near-Infrared-(NIR)-fs-Laser).
- Anregungs- und Emissionslicht können im selben Strahlengang optisch getrennt werden und
- die Größe der zu untersuchenden Objekte kann aufgrund ihres Eigenleuchtens bei ausreichend hohem Kontrast weit unter der Auflösungsgrenze eines Lichtmikroskopes liegen.
Der Grundaufbau der meisten Fluoreszenzmikroskope entspricht dem eines Auflichtmikroskopes. Das zu beobachtende Objekt wird dabei nicht durchstrahlt, sondern durch das Objektiv beleuchtet. (Die korrekte Bezeichnung lautet daher Epifluoreszenzmikroskop). Als Lichtquellen werden in der Regel Quecksilberdampflampen oder Laser eingesetzt. Quecksilberdampflampen emittieren Licht über das gesamte sichtbare Spektrum sowie im ultravioletten Bereich. Die für die Anregung des Fluoreszenzfarbstoffes notwendige Wellenlänge wird mit optischen Filtern isoliert und das einfarbige Licht auf das Objekt geleitet, worauf dieses zu fluoreszieren beginnt. Das emittierte, in der Regel längerwellige Fluoreszenzlicht wird durch das Objektiv gesammelt. Im Strahlengang befindliche Farbteiler trennen anschließend das Fluoreszenzlicht vom anregenden Licht und leiten es in das Okular des Mikroskopes, auf eine Fotokamera (analog oder digital) oder auf einen elektronischen Verstärker (Photomultiplier).
Geschichte
Dass Strukturen unter dem Mikroskop eine Leuchterscheinung zeigen, wenn diese mit kurzwelligem Licht bestrahlt werden, wurde erstmals 1904 von August Köhler beobachtet. Die Fluoreszenzmikroskopie wurde aus dieser Beobachtung abgleitet und bei der Firma Carl Zeiss von August Köhler und Henry Siedentopf entwickelt und ihre Anwendung als "Lumineszenzmikroskopie" am 4. April 1908 von A. Köhler erstmals der Öffentlichkeit anlässlich eines Mikroskopiekurses vorgestellt. Ab etwa 1908 wurden bei Carl Zeiss die ersten Mikroskope zur Anregung von Fluorochromen entwickelt.
Anwendungen
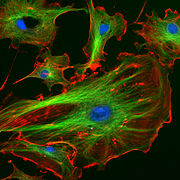 Endothelzellen unter dem Fluoreszenzmikroskop. Die Mikrotubuli sind grün, Aktinfilamente sind rot markiert worden. Die DNA in den Zellkernen wurde mit DAPI angefärbt.
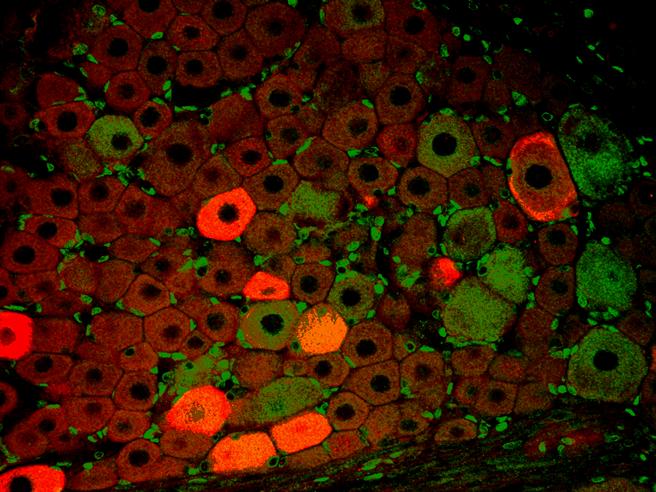
Endothelzellen unter dem Fluoreszenzmikroskop. Die Mikrotubuli sind grün, Aktinfilamente sind rot markiert worden. Die DNA in den Zellkernen wurde mit DAPI angefärbt.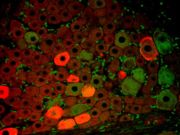 Immunfluoreszenz-Aufnahme im Spinalganglion der Ratte. Zwei verschiedene Proteine wurden mit rot oder grün fluoreszierenden Markern gefärbt.
Immunfluoreszenz-Aufnahme im Spinalganglion der Ratte. Zwei verschiedene Proteine wurden mit rot oder grün fluoreszierenden Markern gefärbt.In der Biologie werden Fluoreszenzmikroskope vielfältig eingesetzt. Im einfachsten Falle werden Zellbestandteile, die von Natur aus fluoreszieren (Autofluoreszenz) sichtbar gemacht. Dies ist vor allem bei Pflanzen möglich, denn Chlorophylle und andere pflanzliche Pigmente haben die natürliche Eigenschaft, zu fluoreszieren. Im Allgemeinen ist diese Eigenschaft aber unerwünscht, denn unspezifische Autofluoreszenz verschiedener Zellbestandteile führt zu starken Hintergrundsignalen. Siehe auch: TIRF
Die wichtigsten Anwendungen basieren auf der spezifischen Färbung einzelner Zellbestandteile, meist bestimmter Proteine. Diese werden zuvor entweder mit Hilfe spezifisch bindender fluoreszenzmarkierter Liganden (z. B. Phalloidin-FITC für Aktin, DAPI für DNA), spezifischer Antikörper (Immunfluoreszenz) oder durch Fusion mit einem fluoreszierenden Protein wie GFP markiert. Aus dem fluoreszenzmikroskopischen Bild können anschließend Rückschlüsse auf die Lokalisation des Proteins in der Zelle gezogen (z. B. im Kern, im Cytoplasma, membrangebunden oder nach außen exportiert) bzw. Zellbestandteile durch ihre spezifischen Proteine visualisiert werden (z. B. Cytoskelett durch Aktin, Mikrotubuli durch Tubulin). Auch Interaktionen von Proteinen untereinander sind beobachtbar. Durch Genexpression eines Markerproteins wie GFP unter Kontrolle eines spezifischen Promotors können auch Zelltypen identifiziert werden, die allein aufgrund ihres lichtmikroskopischen Erscheinungsbild nicht sicher angesprochen werden können. Je nach angewandter Methode ist auch die Verfolgung einzelner Vorgänge in lebenden Zellen möglich.Heute existiert eine breite Palette an Fluoreszenzmarkern, Markierungsmethoden und Mikroskopietechniken.
Spezielle Verfahren der Fluoreszenzmikroskopie
Konfokalmikroskopie
Bei der Konfokalmikroskopie, wie auch bei ihrer speziellen Form der Konfokalen Laser Scanning Mikroskopie (CLSM), gelangt durch den Einsatz einer Lochblende nur Fluoreszenzlicht eines winzigen Teiles der angeregten Probe in das Mikroskop. Das Volumen, aus dem die Fluoreszenz dann ausgelesen wird, wird so auf weniger als ein Femtoliter (0,000 000 000 000 001 Liter) beschränkt. Dies hat den entscheidenden Vorteil, dass Hintergrundsignale ebenfalls fluoreszierender Bestandteile aus höher oder tiefer liegenden Schichten des Präparates ausgeblendet werden können. Auf diese Weise ist Mikroskopieren in mehrschichtigen Präparaten, zum Beispiel in ganzen Blättern oder tierischen Embryos, möglich. Durch Scannen übereinander liegender Schichten und anschließende Rekonstruktion der so gewonnenen Einzelbilder können außerdem dreidimensionale Bilder erstellt werden.
2-Photonen-Mikroskopie
Bei der 2-Photonen-Mikroskopie (siehe auch Multiphotonen-Mikroskopie) wird der Effekt ausgenutzt, dass die Fluoreszenzanregung statt durch Absorption eines energiereichen Photons auch durch die zeitgleiche Absorption zweier energieärmerer Photonen erfolgen kann, d. h. dass statt kurzwelligem Licht (z. B. 375 nm), längerwelliges Licht (z. B. 750 nm) zur Anregung genutzt werden kann. Der 2-Photoneneffekt bedarf einer hohen Photonenflussdichte, wie sie nur im Zentrum eines gepulsten Laserstrahles existiert, d. h. nur in diesem Bereich wird die Probe zur Fluoreszenz angeregt. Im Gegensatz zur Konfokalmikroskopie, wo das Auslesevolumen beschränkt ist, ist hier das Anregungsvolumen limitiert, was zu einer wesentlich schonenderen Behandlung der oft empfindlichen biologischen Proben führt. Zudem hat längerwelliges Laserlicht eine höhere Eindringtiefe in biologische Proben, z. B. Geweben, und führt zu weniger chemischen Bindungsbrüchen als z. B. kurzwelliges Laserlicht (UV). Durch die so etwa verdoppelte Wellenlänge des anregenden Lichtes kommt es hier zu dem scheinbaren Paradoxon, dass die Fluoreszenz kurzwelliger ist als die sie verursachende Anregung. Typische Anregungsquellen sind gepumpte Titan-Saphir-Laser, die im 90 bis 120 fs-Bereich pulsen, und von etwa 750 nm bis 1100 nm durchstimmbar sind (Nahes Infrarot- NIR)
Vertico-SMI-Mikroskopie
Die Vertico-SMI-Mikroskopie (SMI steht für Spatially Modulated Illumination) ist eine optische Nanoskopie, die mit Weitfeldaufnahmen ganze lebende Zellen aufnehmen kann. Es werden Auflösungen bis zu 10 nm in 2D und 40 nm in 3D erreicht. Die Aufnahmegeschwindigkeit bei 3D beträgt hierbei 2 Minuten pro Zelle. Diese Mikroskopie funktioniert mit normalen Fluoreszenzfarbstoffen. Das 3D-Vertico-SMI beruht auf einer Kombination der Lokalisationsmikroskopie SPDM mit strukturierter Beleuchtung SMI.[1]
STED-Mikroskopie
Bei der STED-Mikroskopie (STED steht für Stimulated Emission Depletion) wird eine Auflösung bis zu 50 nm erreicht. Die Anregung erfolgt durch Kombination von UV-Licht mit einem nachfolgenden roten Lichtstrahl. Dabei fallen die angeregten Moleküle an den Rändern wieder in den Normalzustand. Das emittierende Volumen verkleinert sich dadurch und die Auflösung des Mikroskops erhöht sich. Die Proben erleiden keine Strahlenschäden, so dass sich auch lebende Zellen beobachten lassen[2].
TIRF-Mikroskopie
TIRF-Mikroskopie (Total internal reflection fluorescence microscopy) ist eine Methode der Fluoreszenzmikroskopie, um Strukturen zu untersuchen, die sich sehr nahe (ca. 200 nm) an Kontaktoberfläche der Probe befinden. Dadurch ergibt sich ein deutlich höherer Kontrast, da nur wenig Material zur Fluoreszenz angeregt wird, was dadurch erreicht wird, dass man den Objektträger von hinten und in einem Winkel bestrahlt wird, der groß genug ist, damit Totalreflexion auftritt. Da die Strahlung aber dennoch in den Objektträger eindringt, aber dabei gleichzeitig mit zunehmenden Weg immer stärker abgeschwächt wird (sog. evaneszente Welle), erreicht sie nur wenige Schichten fluoreszierenden Materials.
Weblinks
Fußnoten
- ↑ Reymann J, Baddeley D, Gunkel M, Lemmer P, Stadter W, Jegou T, Rippe K, Cremer C, Birk U.: High-precision structural analysis of subnuclear complexes in fixed and live cells via spatially modulated illumination (SMI) microscopy. in: Chromosome Research 2008;16(3):367-82.
- ↑ V. Westphal, S. O. Rizzoli, M. A. Lauterbach, D.Kamin, R. Jahn, S. W. Hell: Video-Rate Far-FieldOptical Nanoscopy Dissects Synaptic Vesicle Movement. in: Science. Washington 320.2008, S.246-249. ISSN 0036-8075


Wikimedia Foundation.