- Albright-Syndrom
-
Fibröse Dysplasie ist eine chronische Fehlbildung des Knochens, bei der der Knochen nicht in seiner üblichen Zellstruktur wächst, sondern geschwulstartige Auswüchse mit unregelmäßiger Zellstruktur bildet. Es handelt sich dabei wie bei allen Dysplasien von Skelett um eine Störung des Knochengewebes, also um einen Gewebsdefekt. Eine alternative Bezeichnung ist Morbus Jaffé-Lichtenstein. In Verbindung mit sichtbaren hormonellen Störungen (Frühreife, Pigmentstörungen der Haut) und starken Verformungen der Kiefer spricht man auch vom McCune-Albright-Syndrom.
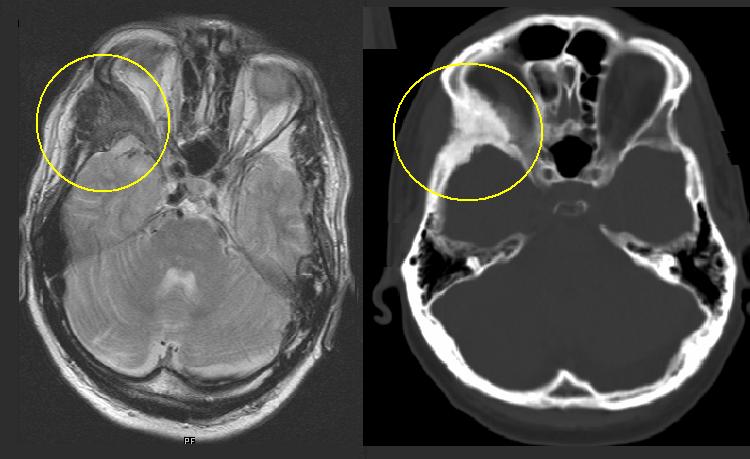
 Fibröse Dysplasie des Jochbeines (markiert). Korrespondierende T2-gewichtete Kernspintomographie (links) und CT (rechts) desselben Patienten.
Fibröse Dysplasie des Jochbeines (markiert). Korrespondierende T2-gewichtete Kernspintomographie (links) und CT (rechts) desselben Patienten.Inhaltsverzeichnis
Definition
Fibröse Dysplasie ist eine seltene Erkrankung des menschlichen Skelettsystems. Sie ist die häufigste Knochenfehlbildung, die im Kindes- und Jugendalter auftritt. Jedoch gibt es auch Erkrankungen im Erwachsenenalter. Bei der fibrösen Dysplasie handelt es sich um eine chronische Störung im Aufbau von neuer Knochenmasse (Störung der Osteoblasten-Differenzierung). Statt strukturiertem und mineralisiertem neuen Knochen wird ein dem Bindegewebe ähnliches, fibröses Knochengewebe gebildet, in dem die faserigen Knochenzellen nicht ausgerichtet sind. Das führt zu einer Auftreibung des Knochens und einem erhöhten Frakturrisiko.
Man unterscheidet Formen der fibrösen Dysplasie, die nur einen Knochen befallen (monostotisch) und Formen, die mehrere Knochen befallen (polyostotisch). Obwohl mehrere Knochen von fibröser Dysplasie gleichzeitig befallen sein können, handelt es sich nicht um eine Krankheit, die sich von einem Knochen zum anderen ausbreitet oder benachbarte Knochen „infiziert“.
Fibröse Dysplasie kann an allen Knochen auftreten. Die häufigsten betroffenen Knochen sind:
- Schädel- und Gesichtsknochen
- Oberschenkelknochen
- Schienbein
- Oberarmknochen
- Rippen
- Hüfte
Symptome/Probleme
Betroffene Knochen wachsen unregelmäßig und entwickeln Deformierungen, die durch die übermäßige fibröse Gewebsvermehrung hervorgerufen werden. Es nimmt zwar das Volumen der Knochen zu, jedoch führt der Verlust der stabilisierenden Binnenstruktur (Spongiosagerüst) zur strukturellen Schwächung und somit zu Belastungsschmerzen und erhöhter Frakturgefahr.
Neben häufigen Knochenbrüchen äußert sich eine fibröse Dysplasie auch in einer Störung des Hormonhaushalts (z. B. Überschuss an Wachstumshormonen), weshalb an fibröser Dysplasie erkrankten Heranwachsenden oft ein beschleunigter Reifungsprozess beobachtet werden kann. Es können auch Pigmentstörungen der Haut („Café-au-lait-Flecken“) oder auch starke Verformungen der Kieferknochen (Cherubismus), verbunden mit den bereits erwähnten hormonellen Störungen (Cushing-Syndrom, McCune-Albright-Syndrom) auftreten. Eine „einfache“ (und zumeist monostotische, aber auch polyostotische) fibröse Dysplasie wird manchmal auch nach dessen Erforschern als Jaffé-Lichtenstein-Syndrom bezeichnet. Jedoch handelt es sich bei fibröser Dysplasie, McCune-Albright-Syndrom und auch Jaffé-Lichtenstein-Syndrom um die gleiche ursächliche Erkrankung.
Seltener kann eine fibröse Dysplasie gemeinsam mit Myxomen der Skelettmuskulatur (Mazabraud-Syndrom) oder Fehlfunktionen von Herz, Leber, Bauchspeicheldrüse, Schilddrüse oder anderen Organen auftreten.
Durch den verstärkten Knochenumbauprozess ist eine fibröse Dysplasie oft mit einem erhöhten Wert der alkalischen Phosphatase im Blut sowie Hydroxyprolin bzw. Desoxypyridinolin im Urin verbunden (was aber umgedreht nicht gleich eine Erkrankung an fibröser Dysplasie bedeuten muss), weshalb diese Werte bei den Betroffenen regelmäßig untersucht werden sollten, um Veränderungen in der Aktivität der fibrösen Dysplasie festzustellen.
Da fibröse Dysplasie sehr stark mit Hormon- und anderen Zellstoffwechselprozessen verbunden ist, kann eine fibröse Dysplasie nach der Pubertät aufhören. Hormonpräparate (wie z. B. die Pille) oder eine Schwangerschaft können einen positiven oder auch negativen Einfluss auf die Weiterentwicklung der fibrösen Dysplasie haben.
Tritt fibröse Dysplasie an Knochen auf, durch die Nerven- oder Blutbahnen führen, wie an der Wirbelsäule oder dem Schädelknochen, so besteht die Gefahr, dass diese Nervenbahnen oder Gefäße eingeklemmt werden.
Obwohl es teilweise erhebliche physische Einschränkungen bei fibröser Dysplasie-Patienten gibt (z. B. häufige Operationen oder Deformationen), so wurde jedoch im Rahmen einer Studie festgestellt, dass in allen wichtigen Bereichen des Lebens, wie soziale Beziehungen, Bildung und Karriere, die Lebensqualität völlig normal ist. Dagegen leiden Eltern von Kindern, die an fibröser Dysplasie erkrankt sind, oft sehr stark unter der Erkrankung. Es sei daher wichtig, die Eltern über die Krankheit aufzuklären und zu versichern, dass Lebenserwartung sowie soziale und emotionale Gesundheit der Kinder nicht beeinträchtigt ist.
Ursachen
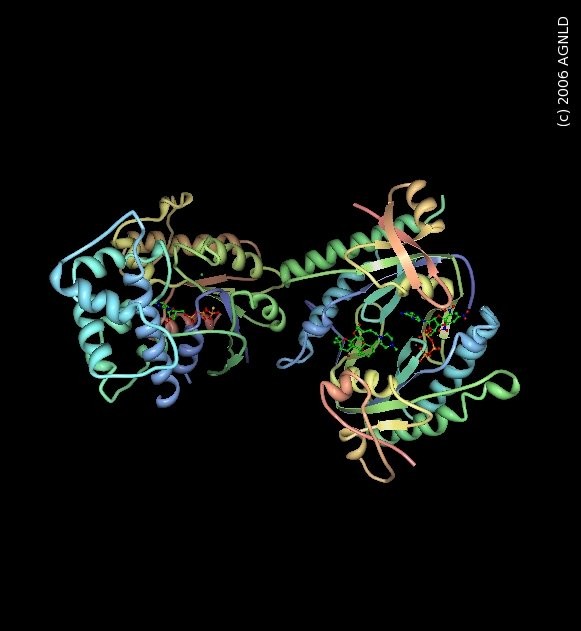
 Komplex des Gs-Proteins (2GVD). Der α-Abschnitt, der bei einer fibrösen Dysplasie verändert ist, befindet sich unten rechts.
Komplex des Gs-Proteins (2GVD). Der α-Abschnitt, der bei einer fibrösen Dysplasie verändert ist, befindet sich unten rechts.Fibröse Dysplasie wird durch eine nichtvererbbare Mutation des G-Proteins ausgelöst (im α-subunit). Die Mutation befindet sich im Gen GNAS (oder auch Gsα-Gen) des 20. Chromosoms. G-Proteine sind im Zell-Stoffwechsel für die Signalweiterleitung extrem wichtig. Durch die Mutation kommt es zu einer Überproduktion des Enzyms Adenylylcyclase, das die Katalyse von ATP zu cAMP steuert (das ist dann der eigentliche Signalübertragunsprozess). cAMP regelt z. B. die Herzfrequenz, Relaxation der glatten Muskulatur, die Wirkung von zahlreichen Hormonen und eben auch die Knochenzellen, die für den Aufbau von Knochen verantwortlich sind (Osteoblasten). Der Grund für diese Mutation ist noch unbekannt. Sie kann bereits im Föten-Stadium während der Schwangerschaft auftreten (dann treten die Symptome bereits im Kindes- und Jugendalter auf) oder auch erst später, nach der Geburt. Da fibröse Dysplasie sehr stark mit Hormon- und anderen Zellstoffwechselprozessen verbunden ist, kann eine fibröse Dysplasie nach der Pubertät aufhören. Hormonpräparate (wie z. B. die Pille) oder eine Schwangerschaft können einen positiven oder auch negativen Einfluss auf die Weiterentwicklung der fibrösen Dysplasie haben.
Der Gendefekt betrifft nur Körperzellen, nicht die Keimzellen (das erste nennt man dann somatische Mutation, dagegen bei Keimzellen gametische Mutation). D. h., der Gendefekt kann nicht an die Nachkommen weitergegeben werden. Findet die Mutation, die fibröse Dysplasie auslöst, während der frühen embryonalen Entwicklung in der Zellmasse statt, kommt es vermutlich zum McCune-Albright-Syndrom; Mutation zu einem späteren Zeitpunkt der embryonalen Entwicklung löst vermutlich die polyostotische fibröse Dysplasie aus. Und eine Mutation nach der Geburt (im Kindes- oder gar Erwachsenenalter) wird für die monostoische fibröse Dysplasie verantwortlich gemacht. Je nachdem, wo die Mutation in der Zellmasse während der embryonalen Entwicklung stattfindet, gibt es später in diesen Körperregionen die Krankheit.
Die Keimzellen entstehen aus Urkeimzellen, die bereits während des Embryowachstums von den Körperzellen getrennt werden (und dann in der Keimdrüse gelagert werden). Eine Vererbung von fibröser Dysplasie ist daher nahezu ausgeschlossen.
Diagnose
Von fibröser Dysplasie befallene Knochenbereiche weisen ein charakteristisches, milchglasartiges und klar begrenztes Erscheinungsbild im Röntgen oder CT auf. Im Zweifel bringt eine Biopsie und Betrachtung einer Knochenprobe unter dem Mikroskop Gewissheit.
Verwechslungsmöglichkeit mit Osteodystrophia deformans (Morbus Paget).
Behandlungsmethoden
Fibröse Dysplasie wird oftmals nur symptomatisch behandelt, das heißt durch
- medikamentöse Schmerzlinderung
- Stabilisierung und Verstärkung eines betroffenen Knochens
- Entfernung von Knochenmaterial, welches die Bewegung einschränkt, Nervenbahnen oder Blutgefäße einzuklemmen droht
- Entfernung von Knochendeformierungen aus kosmetischen Gründen.
Eine medikamentöse Behandlung, die die Knochendeformierung wirksam stoppt, ist noch nicht gefunden, jedoch haben sich Bisphosphonate zur Verlangsamung der fibrösen Dysplasie und Schmerzlinderung bewährt. Es gibt verschiedene Klassen von Bisphosphonaten, die alle die Wirkung der knochenabbauenden Zellen (Osteoklasten) einschränken, und damit die fibröse Dysplasie verzögern und auch Schmerzen lindern können. Am häufigsten finden Pamidronate, Risedronate und Zoledronate Anwendung.
Ein neues Medikament aus der Osteoporose-Medikation mit dem Namen Strontiumranelat könnte künftig Anwendung finden.
Literatur
- R. D. Chapurlat, P. J. Meunier: Fibrous dysplasia of bone. In: Best Practice & Research Clinical Rheumatology. 14, Nr. 2, 2000, S. 385–398. doi:10.1053/berh.1999.0071
- R. Chapurlat: Current pharmacological treatment for fibrous dysplasia and perspectives for the future. In: Joint Bone Spine. 72, Nr. 3, 2005, S. 196-198. doi:10.1016/j.jbspin.2004.08.001
- M. T. Collins, P. Bianco: Fibrous Dysplasia. In: American Society of Bone and Mineral Research, S. 466–470 2003
- M. T. Collins: McCune-Albright Syndrome. In: Orpha.net. 2004
- A. Grauer, K. Abendroth, M. Heller, H.-P. Kruse, H. W. Minne, J. D. Ringe, D. Sabo, A. Schulz, J. Semler: Der Morbus Paget des Knochens: Epidemiologie, Diagnostik und Vorschläge für die Therapie. In: Deutsches Ärzteblatt. 95, Nr. 34-35, 1998, S. A-2021–A-2026
- T. E. Hullar, L. R. Lustig: Paget's disease and fibrous dysplasia. In: Otolaryngologic Clinics of North America. 36, Nr. 4, 2003, S. 707-732
- M. H. Kelly, B. Brillante, H. Kushner, P. Gehron Robey, M. T. Collins: Physical function is impaired but quality of life preserved in patients with fibrous dysplasia of bone. In: Bone. 37, Nr. 3, 2005, S. 338-394. doi:10.1016/j.bone.2005.04.026
- A. I. Leet, C. Chebli, H. Kushner, C. C. Chen, M. H. Kelly, B. A. Brillante, P. G. Robey, P. Bianco, S. Wientroub, M. T. Collins: Fracture Incidence in Polyostotic Fibrous Dysplasia and the McCune-Albright Syndrome. In: Journal of Bone and Mineral Research. 19, Nr. 4, 2004, S. 571-577. doi:10.1359/JBMR.0301262
- P. Mikosch: Die Knochenszintigraphie in der Diagnostik metabolischer Knochenerkrankungen. In: WMW Wiener Medizinische Wochenschrift. 154, Nr. 5-6, 2004, S. 119-126. doi:10.1007/s10354-004-0053-4
- D. Selva, V. A. White, J. X. O'Connell, J. Rootman: Primary bone tumors of the orbit. In: Survey of Ophthalmology. 49, Nr. 3, 2004, S. 328-342. doi:10.1016/j.survophthal.2004.02.011
- L. S. Weinstein, S. Yu, D. R. Warner, J. Liu: Endocrine Manifestations of Stimulatory G Protein α-Subunit Mutations and the Role of Genomic Imprinting. In: Endocrine Reviews. 22, Nr. 5, 2001, S. 675-705
- S. Wallach: Paget's disease and fibrous dysplasia. In: Current Opinion in Rheumatology. 3, Nr. 3, 1991, S. 472-480
- Proteindatenbank PDB
Weblinks
- Umfangreiche Literatursammlung und Informationen zu Verwechslungsmöglichkeiten und über andere seltene Knochenkrankheiten
- Englischsprachige Webseite mit Informationen, Hilfe und Kommunikationsmöglichkeiten zur Fibrösen Dysplasie
- Die Fibrous Dysplasia Foundation (englisch)
- Selbsthilfe-Seite von McCune-Albright-Syndrom Betroffener

Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.