- 18S rRNA
-
Die ribosomale Ribonukleinsäure, abgekürzt rRNA, ist eine Ribonukleinsäure, die in den Ribosomen vorkommt. Die rRNA ist wie die tRNA eine non-coding RNA. Sie trägt somit keine genetische Information, die in Proteine umgeschrieben wird, sondern ist als RNA-Molekül in der Zelle aktiv. Sie ist zusammen mit den ribosomalen Proteinen am Aufbau und der enzymatischen Aktivität des Ribosoms und damit an der Proteinsynthese beteiligt. Die rRNA wird von der so genannten rDNA codiert.
Inhaltsverzeichnis
Übersicht
Ribosomen von Prokaryoten enthalten stets drei unterschiedlich große rRNA-Moleküle, die der Eukaryoten vier. Die Größe der rRNAs wird entweder in Svedberg (S) oder als Anzahl der Nukleinbasen (Nukleotide) im rRNA-Molekül angegeben:
Prokaryoten (Bakterien und Archaeen) Ribosom Untereinheit rRNA Nukleotide 70 S 50 S 23 S 2900 nt 5 S 120 nt 30 S 16 S 1500 nt Eukaryoten (Pflanzen, Tiere, Pilze, Protozoen) Ribosom Untereinheit rRNA Nukleotide 80 S 60 S 28 S 4718 nt 5,8 S 160 nt 5 S 120 nt 40 S 18 S 1874 nt rRNA der Prokaryoten
Die 16S rRNA macht zusammen mit verschiedenen Proteinen ca. 2/3 der Masse der kleineren 30S-Untereinheit der prokaryontischen Ribosomen aus und ist ein wichtiger Bestandteil der Initiationsphase der Translation. Durch Basenpaarung bindet das 3'-Ende der 16S rRNA an die Shine-Dalgarno-Sequenz der mRNA. Dadurch wird das Startcodon der mRNA in die richtige Position im Ribosom gebracht. Diese Position heißt P-Stelle.
rRNA der Eukaryoten
Die 45S Prä-rRNA wird im Nucleolus von der RNA-Polymerase I transkribiert. Sie wird prozessiert zu 18S, 5,8S und 28S rRNAs. Zusammen mit snoRNAs und Proteinen ergeben diese den 80S Partikel. Zu diesem gelangt die 5S rRNA welche durch RNA-Polymerase III extranucleolär transkribiert wurde. Es entstehen die nichtmaturierten 40S(aus 18S rRNA und 33 Proteinen) und 60S(aus 5S, 5,8S und 28S rRNAs und 50 Proteinen) Untereinheiten. Sie werden aus dem Zellkern transportiert und sind jetzt maturierte 40S und 60S Untereinheiten welche zusammen ein Ribosom bilden.
rRNA und Phylogenetik
Ribosomale RNA erlangte in den letzten Jahrzehnten enorme Bedeutung als Werkzeug zur Aufklärung der Stammesgeschichte, Evolution des Lebens und der Erforschung verwandtschaftlicher Beziehungen unter den Organismen. Die Analyse der rRNA ist heute eine anerkannte Methode zur Einordnung einer Art in den universellen Stammbaum des Lebens und zur Ermittlung der nächst verwandten Arten.
rRNA als idealer molekularer Marker
Ribosomale RNA war wahrscheinlich bereits Bestandteil der ersten lebenden Einheiten auf der Erde und damit der Vorfahren aller heute lebenden Organismen. Sie gehört zur Grundausstattung jeder heute lebenden Zelle. Gleichzeitig hat sie in allen Organismen die gleiche Funktion und die Gene der rRNA unterliegen wahrscheinlich nur selten einem horizontalen Gentransfer. Man geht deshalb davon aus, dass die rRNA-Moleküle in allen Organismen mit vergleichbarer Geschwindigkeit evolvieren und nicht nur die Entwicklungsgeschichte des jeweiligen rRNA-Genes sondern die eines gesamten Organismus widerspiegeln. Sie gelten als ideale "molekulare Chronometer", mit deren Hilfe sich verwandtschaftliche Beziehungen unter allen Organismen rekonstruieren lassen.
RNA ist jedoch ein instabiles Molekül und deren Analyse technisch aufwendig. DNA ist wesentlich stabiler als RNA und einfacher zu handhaben. Deshalb arbeitet man in der Praxis fast immer mit den Genen der rRNA, also der rDNA und leitet hiervon die Sequenz der rRNA ab.
Informationseinheiten
Ribosomale RNA besteht - wie jede RNA und DNA auch - aus einer Kette hintereinander angeordneter Nukleotide. Die Abfolge dieser Nukleotide - in einer festgelegten Richtung gelesen - ergibt die Nukleotidsequenz des rRNA-Moleküls. Diese Sequenz enthält die eigentliche phylogenetische Information. Jede einzelne Base dient dabei als Informationseinheit. Aufschluss über verwandtschaftliche Beziehungen liefern die Unterschiede beim Vergleich zweier rRNA-Sequenzen.
Auswahl geeigneter rRNA-Moleküle
Die kleinen 5- und 5,8S rRNAs liefern aufgrund ihrer geringen Nukleotidanzahl zu wenig phylogenetische Information, die Analyse der großen rRNAs ist schwieriger. Deshalb arbeitet man vorwiegend mit 16S rRNAs bei Prokaryonten und 18S rRNAs bei Eukaryonten. Diese Sequenzdatenbanken sind derzeit die umfangreichsten.
Auswahl geeigneter Sequenzabschnitte
rRNA-Moleküle verfügen über Abschnitte, die unterschiedlich stark konserviert sind. Hoch konservierte Bereiche sind in ihrer Struktur für die Funktion der Ribosomen unverzichtbar. Man findet sie daher in allen Organismen nahezu unverändert. Solche Sequenzabschnitte bilden oft doppelsträngige Sekundärstrukturen mit ringförmigen loops. Veränderungen in der Basenabfolge sind für den betroffenen Organismus meist nachteilig oder tödlich. Entsprechend werden solche Mutationen in einer Population nur sehr selten fixiert.
Die Basensequenz anderer Abschnitte ist variabler. Mutationen können hier zum Beispiel durch Veränderungen an ribosomalen Proteinen kompensiert werden. Im Laufe der Zeit können sich solche Mutationen in einer Population oder einer ganzen Art durchsetzen.
Schließlich existieren auch Abschnitte, deren Sequenz nahezu beliebig variieren kann, ohne die Funktionsweise der Ribosomen zu beeinträchtigen. Diese Regionen unterliegen kaum einer Selektion und verändern sich um den Faktor Tausend mal schneller, als konservierte Abschnitte.
Die grundlegende Annahme für die Analyse verwandtschaftlicher Verhältnisse ist, dass die Anzahl fixierter Mutationen, also Veränderungen in der Basensequenz, proportional mit der Zeit zunimmt. Zur Unterscheidung nah verwandter Organismen eignen sich hoch variable Regionen der rRNA, denn nur hier finden sich informative Sequenzunterschiede. Entfernt verwandte oder unbekannte Organismen werden hingegen anhand stärker oder hoch konservierter Sequenzabschnitte analysiert.
Sequenzanalyse
Sind zwei Arten sehr eng miteinander verwandt, so haben sie stammesgeschichtlich erst vor kurzer Zeit eine eigenständige Entwicklung genommen. In dieser kurzen Zeitspanne sind kaum Veränderungen in den rRNA-Sequenzen beider Arten aufgetreten. Die Sequenzen können aneinander ausgerichtet werden (Alignment), sie sind quasi deckungsgleich. Einzelne Sequenzunterschiede können dann relativ leicht erkannt werden.
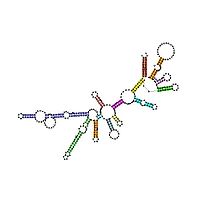 Sekundärstruktur der 5'-Domäne einer rRNA mit charakteristischen Schleifen (loops). Quelle: Rfam-Datenbank, RF00177
Sekundärstruktur der 5'-Domäne einer rRNA mit charakteristischen Schleifen (loops). Quelle: Rfam-Datenbank, RF00177Beispiel:
- Sequenz von Art A: ...UAGCUAAGGUGAACAC...
- Sequenz von Art B: ...UAGCUAACGUGAUCAC...
Vergleicht man jedoch die rRNAs von zwei entfernt verwandten Arten, ist das Alignment schwierig. Die Sequenzen sind zu verschieden, um sie ohne weiteres aneinander ausrichten zu können. Nur die hoch konservierten funktionellen Abschnitte geben noch Aufschluss, wie die Sequenzen zusammen passen. Weil aber auch konservierte Bereiche in ihrer Sequenz variieren, hilft oft eine Vorhersage möglicher Sekundärstrukturen (loops), die - wie oben angedeutet - funktionell konserviert sind.
Das korrekte Ausrichten der Sequenzen ist der wichtigste und oft auch schwierigste Teil der Sequenzanalyse. Es ist unbedingt notwendig, die Sequenzabschnitte der einen Art mit genau den entsprechenden Abschnitten der anderen Art zu vergleichen. (Sind beide Sequenzen gegeneinander verschoben, wird das Ergebnis der Analyse verfälscht. Dies geschieht bei rRNAs von unbekannten Arten relativ leicht, denn rRNA-Moleküle verschiedener Arten können unterschiedlich lang sein und ganze Abschnitte fehlen.)
Mit Hilfe mathematischer Verfahren, die auch Reversionen, also Rückmutationen berücksichtigen, wird aus den erkannten Sequenzunterschieden ein "phylogenetischer Abstand" errechnet, und dieser meist in Form eines Dendrogrammes oder "Stammbaumes" dargestellt. Je größer die Sequenzunterschiede, desto größer der phylogenetische Abstand und desto länger die "Äste" im Dendrogramm.
Anwendungen und Erkenntnisse
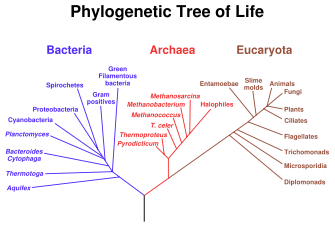 Universeller Stammbaum des Lebens, auf Basis der rRNA-Sequenzen erstellt.
Universeller Stammbaum des Lebens, auf Basis der rRNA-Sequenzen erstellt.Die auf Basis der ribosomalen RNA entwickelten Stammbäume gelten inzwischen als zuverlässig und die meisten der hiermit errechneten Verwandtschaftsbeziehungen wurden auch mit anderen Methoden bestätigt. Dennoch kann die Anwendung der rRNA-Methode nicht allein für die korrekte Einordnung eines Organismus herangezogen werden. Die errechnete Position im Stammbaum muss stets mit anderen Methoden bestätigt werden. Hierzu zählen nach wie vor auch morphologische und physiologische Merkmale. So ist es beispielsweise nicht möglich, allein auf Basis einer rRNA-Analyse eine neue Art zu definieren.
Große Bedeutung hat die rRNA-basierte Phylogenetik bei Mikroorganismen, denn Einzeller sind anhand morphologischer und physiologischer Merkmale allein schwer einzuordnen. Hier bietet die Analyse der ribosomalen RNA eine schnelle und zuverlässige Ergänzung. Anhand empirischer Daten geht man heute davon aus, dass Bakterien, deren 16S rRNA-Sequenzen zu 97 - 98 % übereinstimmen, einer Art zugerechnet werden können.
Man hat aus verschiedenen Umweltproben (zum Beispiel Wasser, Boden oder Klärschlamm) DNA isoliert und hieraus rRNA-Sequenzen bestimmt. In einem Gramm Waldboden fand man so zum Beispiel rRNA-Gene von etwa 13.000 (!) verschiedenen „Arten“. Vergleicht man diese Sequenzen mit denen von kultivierbaren und daher bekannten Mikroorganismen, kann man abschätzen, dass wir heute nur 1 - 5 % aller Mikroorganismen überhaupt kennen. Von der Existenz der übergroßen Mehrheit aller Bakterien und Archaeen weiß man nur durch ihre rRNA-Sequenzen, ohne eine Vorstellung davon zu haben, wovon sie leben und welche Rolle sie in der Natur spielen.
Alle bisher gültig beschriebenen (das heißt kultivierbaren) Bakterien werden - je nach Autor - derzeit in 26 Phyla oder Stämme eingeordnet. Jedoch verteilt sich die übergroße Mehrheit aller Bakterien auf nur wenige Stämme, zum Beispiel auf die Proteobacteria, Firmicutes und Actinobacteria. Die meisten Phyla werden hingegen nur durch einen oder wenige kultivierbare Vertreter repräsentiert (zum Beispiel Acidobacteria), obwohl man weiß, dass diese Gruppen viel mehr Vertreter umfassen müssen.
26 weitere Phyla werden nur mit Hilfe von aus Umweltproben isolierten rRNA-Sequenzen postuliert, ohne bisher einen Vertreter kultiviert und charakterisiert zu haben.
Die wichtigste Konsequenz aus der Anwendung der rRNA-basierten Phylogenetik war bisher aber die Einteilung aller Organismen in die drei Domänen der Bakterien, Archaeen und Eukaryonten. Außerdem konnte die Endosymbiontentheorie bestätigt werden. Aber auch die derzeit aktuelle Einteilung der Urmünder (Protostomia), der artenreichsten Tiergruppe, in Häutungstiere (Ecdysozoa, u. a. Insekten, Fadenwürmer) und Lophotrochozoen (Lophotrochozoa, u. a. Weichtiere, Ringelwürmer) ist vor allem anhand von Untersuchungen der 18S-rRNA der Ribosomen entwickelt worden.
Molekulare Marker
- siehe Hauptartikel: Marker
Andere molekulare Marker sind zum Beispiel
- der Elongationsfaktor Tu (EfTu)
- das Gen der Untereinheit I der Cytochrom c Oxidase
- das Gen des Cytochrom b der Cytochrom c Reduktase
- die Gene für die ATP-Synthetase
- Hitzeschockproteine oder
- ITS für sehr eng verwandte Eukaryonten oder zur Unterscheidung einzelner Stämme
Siehe auch
- Sequenz von Art A: ...UAGCUAAGGUGAACAC...

Wikimedia Foundation.