- Geschichte der Kernspaltung
-
 Kaiser-Wilhelm-Institut für Chemie (heute: Otto-Hahn-Bau der Freien Universität Berlin) 52.44866613.342959Koordinaten: 52° 26′ 55,2″ N, 13° 20′ 34,65″ O
Kaiser-Wilhelm-Institut für Chemie (heute: Otto-Hahn-Bau der Freien Universität Berlin) 52.44866613.342959Koordinaten: 52° 26′ 55,2″ N, 13° 20′ 34,65″ ODie Entdeckung der Kernspaltung im Dezember 1938 am Kaiser-Wilhelm-Institut für Chemie in Berlin ist eines der bedeutendsten und folgenreichsten Ereignisse in der Geschichte der Naturwissenschaften. Bei der Bestrahlung von Uran mit Neutronen entstanden Spaltprodukte des Urans, u. a. das zuerst nachgewiesene Barium. Dieses entscheidende Ergebnis eines kernphysikalischen und radiochemischen Experiments von Otto Hahn wurde durch exzellent durchdachte chemische Analysen, ausgeführt von seinem Assistenten Fritz Straßmann, gefunden und bewiesen. In interdisziplinärer Zusammenarbeit wurde dieses unerwartete Ergebnis im Januar 1939 durch Lise Meitner und Otto Frisch kernphysikalisch erklärt.
Den Ausgangspunkt bildeten die Versuche von Enrico Fermi, der 1934 Uran mit Neutronen bestrahlt hatte. In jahrelanger Arbeit versuchten Hahn, Meitner und Straßmann, die dabei beobachteten Vorgänge aufzuklären. Unabhängig hiervon widmete sich von 1937 an auch eine Arbeitsgruppe um Irène Joliot-Curie am Radium-Institut in Paris dem gleichen Thema. Anfangs verfolgten alle Arbeitsgruppen die Hypothese, dass bei den Bestrahlungen schwerere Elemente als Uran (sogenannte Transurane) entstehen. Im Dezember 1938 kam es zu einem unerwarteten Ergebnis: Hahn und Straßmann wiesen mit Hilfe spezieller chemischer Trenn- und Analysenverfahren nach, dass es sich bei den beobachteten Reaktionsprodukten um in der Natur nicht vorkommende radioaktive Bariumisotope handelte; es kam bei den Versuchen offenbar zu einem „Zerplatzen“ des Atomkerns, das sie sich nicht erklären konnten.
Lise Meitner, die Deutschland aufgrund ihrer jüdischen Abstammung im Sommer 1938 verlassen musste, gelang im Januar 1939 gemeinsam mit ihrem Neffen Otto Frisch die kernphysikalische Deutung der chemischen Analysenergebnisse. Ihr Modell beschrieb den Urankern als elektrisch geladenen Flüssigkeitstropfen, der durch das Einfangen des Neutrons so in Schwingungen versetzt wurde, dass er sich in zwei annähernd gleich große Fragmente teilte, wobei eine hohe Energie freigesetzt wurde. Frisch gab der bisher unbekannten Kernreaktion den Namen „nuclear fission“ (Kernspaltung). Die Veröffentlichung dieser Ergebnisse im Februar 1939 löste eine außerordentliche Resonanz unter den Naturwissenschaftlern aus, weil die Kernspaltung eine neue Energiequelle von bisher unbekannter Größenordnung erschloss, die Kernenergie.
Vorbemerkungen zur Nomenklatur und Schreibweise
Im Rahmen dieses Artikels werden die in der Kernchemie und Kernphysik in den dreißiger Jahren üblichen Bezeichnungen verwendet, auch wenn sie heute nicht mehr gebräuchlich oder durch neue Begriffe ersetzt sind, so z. B. Atomgewicht durch Atommasse. Für radiochemische Präparate werden die in den damaligen Protokollen und Veröffentlichungen benutzten Bezeichnungen wie „Aktivität“, „Körper“ oder „β-Strahler“ beibehalten, oft in Zusammensetzungen wie „13-min-Aktivität“ oder „3,5-Stunden-Körper“, wobei mit der jeweiligen Zeitangabe die Halbwertszeit (HWZ) gemeint ist. Die dabei verwendeten Zeiteinheiten sind s (Sekunden), m (Minuten), h (Stunden), d (Tage) und a (Jahre).
Auch die damals üblichen Benennungen einzelner radioaktiver Nuklide, wie z. B. Mesothorium 1 (MsTh1) oder Thorium X (ThX), die beide keine Thorium-, sondern Radiumisotope sind, werden beibehalten, jedoch durch die heutige Schreibweise ergänzt.
Für das Kaiser-Wilhelm-Institut für Chemie in Berlin wird die Abkürzung KWI verwendet.
Der Stand der Forschung Anfang der 1930er Jahre
Das Periodensystem der Elemente
Die Arbeitsgruppen, die sich nach der Entdeckung Fermis an der Suche nach den „Transuranen“ beteiligten, standen vor dem Problem, eine durch die Neutronenbestrahlung in nur unwägbaren Mengen gebildete, neue radioaktive Substanz zu identifizieren. Hierbei spielt das periodische System der chemischen Elemente eine wichtige Rolle. Da es in seiner damaligen Form jedoch von der heute üblichen Anordnung der Elemente an einigen Stellen abweicht, wird es hier (vereinfacht) dargestellt und erläutert.
-
H
1He
2Li
3Be
4B
5C
6N
7O
8F
9Ne
10Na
11Mg
12Al
13Si
14P
15S
16Cl
17Ar
18K
19Ca
20Sc
21Ti
22V
23Cr
24Mn
25Fe
26Co
27Ni
28Cu
29Zn
30Ga
31Ge
32As
33Se
34Br
35Kr
36Rb
37Sr
38Y
39Zr
40Nb
41Mo
42Ma
43Ru
44Rh
45Pd
46Ag
47Cd
48In
49Sn
50Sb
51Te
52J
53X
54Cs
55Ba
56La
57Hf
72Ta
73W
74Re
75Os
76Ir
77Pt
78Au
79Hg
80Tl
81Pb
82Bi
83Po
84?
85Rn
86?
87Ra
88Ac
89Th
90Pa
91U
92?
93?
94?
95?
96Lantha-
niden:La
57Ce
58Pr
59Nd
60?
61Sm
62Eu
63Gd
64Tb
65Dy
66Ho
67Er
68Tm
69Yb
70Cp
71
- Periodensystem der Elemente (1938)
- Die chemischen Elemente werden in der Reihenfolge ihrer Ordnungszahl (Kernladungszahl) von links nach rechts in Tabellenform angeordnet, wobei in periodischen Abständen, die teils gleich sind, mehrmals aber auch sprunghaft zunehmen, jeweils eine neue Reihe der Tabelle begonnen wird. Die dann untereinander stehenden Elemente gehören einer „Gruppe“ an, sie zeigen ein ähnliches chemisches Verhalten, man bezeichnet sie deshalb auch als chemisch „homologe“ Elemente.
- Die Elemente 61, 85 und 87 sind damals noch unbekannt, die Elemente 43 (Masurium) und 71 (Cassiopeium) heißen heute Technetium bzw. Lutetium. Für das Element Jod ist damals das Symbol J (heute I) üblich, für Xenon das Symbol X (heute Xe).
- Die bei der Neutronenbestrahlung des Urans möglicherweise entstehenden beziehungsweise erwarteten „Transurane“ wurden den damaligen Vorstellungen entsprechend in die noch freien Felder des periodischen Systems rechts vom Uran eingeordnet (die Abbildung zeigt diese für die Elemente 93 bis 96 damals vorgesehenen Plätze). Demnach wurde also das Element 93 für homolog zu Rhenium, Masurium und Mangan gehalten, Element 94 wäre dann ein homologes Element zu Eisen, Ruthenium und Osmium usw. Daher rühren auch die damals üblichen und in den zitierten Veröffentlichungen benutzten Bezeichnungen Eka-Rhenium, Eka-Osmium, Eka-Iridium und Eka-Platin für diese noch unbekannten Elemente 93-96.
- Erst Jahre später stellte sich heraus, dass diese Anordnung bereits für Actinium und die ihm nachfolgenden Elemente (einschließlich den Transuranen) nicht zutrifft, sie gehören zur Gruppe der Actinoiden. Die 1938 erwartete chemische Ähnlichkeit zwischen Element 93 und Rhenium war also ein Trugschluss, ebenso die zwischen Element 94 und Eisen.
- Inzwischen haben die chemischen Elemente mit den Ordnungszahlen 93 bis 96 folgende Namen erhalten:
- Von allen sind jeweils mehrere (radioaktive) Isotope bekannt.
Die Entdeckung der künstlichen Radioaktivität
Bis zum Jahre 1933 verstand man unter radioaktiven chemischen Elementen nur die in der Natur vorkommenden „natürlich" radioaktiven Elemente. Dies sind die chemischen Elemente mit den hohen Kernladungszahlen 84 bis 92 und die bei ihrem Zerfall gebildeten radioaktiven Tochtersubstanzen. Daneben gibt es in der Natur noch einige leichtere chemische Elemente wie z. B. Kalium und Rubidium, bei denen einzelne Isotope radioaktiv sind; auch deren natürliche Radioaktivität war damals bereits bekannt.
Dem Ehepaar Irène Joliot-Curie und Frédéric Joliot gelang es 1934, Atomkerne nicht radioaktiver Elemente durch die Bestrahlung mit Alphateilchen aus solchen natürlichen Strahlenquellen – also in heutiger Sprechweise durch Kernreaktionen – in ein benachbartes chemisches Element umzuwandeln und „radioaktiv zu machen“. Im Gegensatz zu den natürlich radioaktiven Isotopen bezeichnete man solche Isotope als „künstlich radioaktiv“. Die Elementumwandlung beschränkte sich allerdings auf die leichteren chemischen Elemente, da bei schwereren Elementen deren hohe positive Kernladung das Eindringen des ebenfalls positiv geladenen α-Teilchens in den Atomkern verhinderte.
Für die Entdeckung dieser künstlichen Radioaktivität erhielten Joliot-Curie 1935 den Nobelpreis für Chemie.
Die Neutronenbestrahlung chemischer Elemente durch Fermi (1934)
In Rom setzte Enrico Fermi in den Jahren 1934/35 die Versuche von Joliot-Curie zur Herstellung künstlich radioaktiver Elemente fort. Zur Bestrahlung verwendete er jedoch das von James Chadwick im Jahre 1932 entdeckte, elektrisch ungeladene Neutron, das im Gegensatz zu den von Curie verwendeten α-Teilchen auch in Atomkerne mit hoher Kernladungszahl ungehindert eindringen kann. An 68 der damals bekannten 89 chemischen Elemente wurden die Bestrahlungen durchgeführt, bei 47 Elementen wurde eine Umwandlung beobachtet, die bei schweren Elementen immer nach folgendem Schema ablief: Das auf den Atomkern auftreffende Neutron wird in den Kern aufgenommen, es entsteht ein Isotop des bestrahlten Elements mit einer um eine Einheit höheren Massenzahl. Dieser Atomkern ist instabil (radioaktiv) und zerfällt unter Aussendung eines β−-Teilchens (Elektron) in ein stabiles Isotop des nächsthöheren Elements; also zum Beispiel:
-
- n = Neutron; Ag = Silber; Cd = Cadmium; β− = Betastrahlung
- 24 s = Halbwertszeit (HWZ) des radioaktiven Silberisotops 110Ag
Fermi findet bei diesen Versuchen, dass die radioaktive Strahlung der Proben dann besonders intensiv ist, wenn sich zwischen Neutronenquelle und Probe eine dicke Schicht aus wasserstoffhaltiger Substanz befindet (z. B. Wasser oder Paraffin), in heutiger Sprechweise ein Moderator. Durch häufige Stöße der Neutronen mit den Wasserstoffkernen (Protonen) wird ihre Geschwindigkeit erheblich abgebremst. Die Wahrscheinlichkeit, dass solche langsamen („thermischen“) Neutronen von einem Atomkern der Probe eingefangen werden, ist wesentlich größer als bei ungebremsten Neutronen.
In der zitierten Literatur wird diese Methode mit „verstärkter“ Bestrahlung bezeichnet.
Die unerkannte Kernspaltung des Urans bei der Suche nach Transuranen
In der Folgezeit wird von Fermi in Rom versucht, durch Bestrahlung von Uran (dem damals schwersten bekannten Element im periodischen System mit der Ordnungszahl 92) mit Neutronen Transurane herzustellen und in Berlin und Paris versucht, dies mit Hilfe der damaligen Annahmen über die chemischen Verwandtschaften dieser noch unbekannten und in der Natur nicht vorkommenden Elemente nachzuweisen.
Entsprechend dem oben für das Silber angegebenen Schema müsste diese Reaktion ablaufen nach
und somit zu einem neuen Element mit der Ordnungszahl 93 führen.
Uran-238 reagiert mit Neutronen zwar nach diesem Schema, aber diese Reaktion wird völlig überlagert von der Spaltung des damals noch unbeachteten Uran-235, dessen zahlreichen Spaltprodukte für Transurane gehalten und als solche beschrieben wurden, wie aus den Folgeartikeln ersichtlich ist. Weil Uran mit Neutronen überwiegend anders reagiert als angenommen und die vermuteten Verwandtschaftsbeziehungen gar nicht gelten, war der Weg zur Aufklärung der Sachverhalte langwierig und mühsam.
Was wirklich geschah:
Dies ist nur ein Beispiel von mehr als 100 vorkommenden Spaltungen von Uran-235, und zwar dasjenige mit dem von Hahn und Straßmann Ende 1938 chemisch nachgewiesenen Barium. Dass eine Kernspaltung vorlag, wurde in den Tagen zwischen Weihnachten 1938 und Neujahr 1939 von Frisch und Meitner bestätigt, dass diese Spaltung aber nur das Uranisotop 235 betrifft, wurde erst 1939 von Nils Bohr erkannt.
Die Neutronenbestrahlung von Uran durch Fermi 1934
Bei der Bestrahlung von Uran findet Fermi anstatt einer neuen Substanz gleich fünf radioaktive Produkte, mit 10 s, 40 s, 13 min, 40 min und etwa 24 h Halbwertszeit. Einen dieser Körper, den mit einer HWZ von 13 Minuten, versucht Fermi chemisch zu identifizieren. Er kann dabei eine Zugehörigkeit zum Uran oder einem seiner leichteren Nachbarelemente ausschließen und äußert deshalb die Vermutung, dass die Ordnungszahl des Elementes größer als 92 sein könnte. Für Element 93 spricht die Tatsache, dass der Körper sich ähnlich wie die für homolog gehaltenen Elemente Mangan und Rhenium verhält, aber auch die Möglichkeit für eine Ordnungszahl 94 oder 95 will Fermi nicht ausschließen, da er annimmt, dass die chemischen Eigenschaften dieser Elemente denen von Element 93 wohl ziemlich ähnlich sind.[1]
Die Bezeichnung „Transurane“ für diese neuen Elemente wird von Fermi nicht verwendet, sie hat sich erst später eingebürgert. Weitere Versuche zur Aufklärung des Reaktionsablaufs und zur chemischen Identifizierung der Umwandlungsprodukte werden von dem Physiker Fermi nicht vorgenommen. 1938 wird ihm für seine Entdeckungen der Nobelpreis für Physik verliehen, nach der Preisverteilung in Stockholm kehrt er nicht in das faschistische Italien zurück, sondern emigriert mit seiner Frau in die USA.
Die Neutronenbestrahlung von Uran durch das KWI für Chemie in Berlin 1934–1938
Das einzige Institut in Deutschland, das sich in dieser Zeit mit radiochemischen und zugleich auch kernphysikalischen Fragen befasste, war das Kaiser-Wilhelm-Institut für Chemie in Berlin. Der damalige Direktor des Instituts, dem gleichzeitig auch die chemisch-radioaktive Abteilung unterstand, war der Radiochemiker Otto Hahn, die Leiterin der physikalisch-radioaktiven Abteilung die Physikerin Lise Meitner. Beide kannten sich seit 1907, beide hatten sich an der Universität in Berlin habilitiert, beide waren (1910 bzw. 1926) zum Professor bzw. zur außerordentlichen Professorin ernannt worden. 1933 allerdings war Lise Meitner die Lehrbefugnis wegen ihrer jüdischen Abstammung entzogen worden, als Österreicherin konnte sie jedoch ihre Arbeit am (nicht staatlichen) KWI zunächst ungehindert fortsetzen.
In der Hahnschen Abteilung arbeitete weiterhin der Chemiker Fritz Straßmann, der 1929, nach seiner Promotion, als Stipendiat eingestellt worden war und 1935 eine Assistentenstelle am Institut erhalten hatte.
Fermis Beobachtungen bei der Neutronenbestrahlung von Uran werden gegen Ende des Jahres 1934 am KWI in Berlin von diesem Team aufgegriffen. Sie waren der Anlass der Suche nach Transuranen, denn der Physiker Fermi hatte die 5 Reaktionsprodukte nicht chemisch identifizieren können; möglich war dies nur in den radiochemischen Laboratorien von Berlin und Paris.
So beginnt damit in Berlin eine Suche nach Transuranen, die volle vier Jahre andauert und zur Entdeckung der Kernspaltung führt.
Natürliches Uran und seine Folgeprodukte
Uran, das in der Natur hauptsächlich in Form von Pechblende (U3O8) gefunden wird, besteht aus den Isotopen 238U (99,28 %), 235U (0,715 %) und 234U (0,005 %). Alle drei Isotope sind α-Strahler und zerfallen in langen Zerfallsreihen von α- und β-Strahlern, bis schließlich beim Blei (206Pb bzw. 207Pb) stabile Endzustände erreicht sind (vgl. Zerfallsreihe sowie Uran-Radium-Reihe und Uran-Actinium-Reihe). Chemisch reines Uran enthält nur noch die Anfangsglieder, nämlich die drei Uranisotope und die sich rasch neu bildenden Folgeprodukte des Uran-238, das Thoriumisotop UX1 und das Protactiniumisotop UX2:
Wird eine handelsübliche Uranverbindung mit Neutronen bestrahlt, so ist bei den schwachen Neutronenquellen der damaligen Zeit die Aktivität des entstehenden Reaktionsproduktes viel schwächer als die Aktivität der beiden nachgebildeten natürlichen Folgeprodukte Thorium-234 und Protactinium-234. Man muss deshalb unmittelbar vor der Neutronenbestrahlung diese beiden störenden Folgeprodukte abtrennen (das Uran „reinigen“).
Vorgereinigtes Uran
Die Vorreinigung geschieht durch den weiter unten beschriebenen Vorgang einer Mitfällung der Folgeprodukte an Eisenhydroxid. Das hierbei in Lösung bleibende Uran wird dann mit Ammoniak als Ammoniumdiuranat (NH4)2U2O7 (Ammoniumpyrouranat) ausgefällt, abfiltriert und getrocknet. Unmittelbar nach dieser Abtrennung des Th- und Pa-Isotops ist das Uran ein reiner Alphastrahler, die beiden Isotope werden jedoch sofort wieder nachgebildet. Sie haben allerdings nach 24,1 Tagen erst 50 % ihrer Gleichgewichtsaktivität erreicht, man kann also mit dem gereinigten Uranpräparat einige Tage arbeiten (bestrahlen, messen, analysieren), ohne eine wesentliche Beeinflussung durch die beiden β-strahlenden Tochtersubstanzen befürchten zu müssen.
Die Bestrahlungsdauer von Uran
Um trotz der Reinigung des Urans zu hinreichend messbaren Aktivitäten zu kommen, müssen größere Mengen (10 bis 100 Gramm Ammoniumdiuranat) bestrahlt werden. Allgemein nimmt die Menge und damit die Aktivität des Reaktionsproduktes mit steigender Dauer der Neutronenbestrahlung zu; sie nähert sich jedoch einem Endwert, wenn die Bestrahlungsdauer z. B. dem Zehnfachen der Halbwertszeit des neuen radioaktiven Körpers entspricht. Bestrahlt man nur für die Dauer einer HWZ, so hat die Aktivität 50 % dieses Endwertes erreicht, bei zwei Halbwertszeiten 75 %, bei drei HWZ 87,5 % usw. Eine Bestrahlungsdauer von mehr als 3 Halbwertszeiten bringt also kaum noch eine Aktivitätserhöhung und ist deshalb wenig sinnvoll.
Hat das Reaktionsprodukt jedoch eine HWZ von Tagen oder Wochen, so muss man sich notfalls mit einer Bestrahlungsdauer begnügen, die noch kürzer als z. B. eine Halbwertszeit ist und die geringere Aktivität des neuen Körpers in Kauf nehmen.
Die Bestrahlungs- und Messapparatur
Die damals verwendete chemische und physikalische Ausrüstung befindet sich heute im Deutschen Museum in München. Die Apparaturen und Geräte, die im KWI in verschiedenen Räumen untergebracht sein mussten, sind im Museum auf einem Tisch zusammengestellt; soweit es sich um radioaktive Substanzen handelt, sind sie durch Attrappen ersetzt. Verglichen mit den heute (2006) zur Verfügung stehenden Messgeräten waren die damaligen Einrichtungen äußerst einfach, wenn nicht gar primitiv.
Die Neutronenquellen zur Bestrahlung des Urans sind pulverförmige Gemische aus Beryllium oder Berylliumsulfat und jeweils etwa 100 mg Radiumsulfat, luftdicht in Glasröhrchen eingeschmolzen.
Wenn die sechs damals vorhandenen Quellen nicht zur Bestrahlung mehrerer Proben benötigt werden, können sie gemeinsam zur Bestrahlung nur einer Probe verwendet werden und so die Aktivität des Reaktionsproduktes erhöhen. Wenn das Uran mit langsamen (thermischen) Neutronen bestrahlt werden soll, befinden sich Uran und Quellen in einem allseits geschlossenen Paraffinblock, der mit Bohrungen zur Aufnahme der Neutronenquellen und des Urans versehen ist.
Nach der Bestrahlung wird die Uranprobe in Wasser oder Säure gelöst und die gebildete radioaktive Substanz vom Uran abgetrennt. Dies geschieht in einem der radiochemischen Laboratorien des KWI mithilfe spezieller chemischer Trennmethoden, die für kurzlebige Körper hinreichend schnell ablaufen müssen. Von den hierzu benötigten chemischen Geräten ist auf dem Arbeitstisch lediglich eine Saugflasche mit aufgesetzter Nutsche und einem runden glatten Filter von 3 cm Durchmesser zu sehen, auf dem die radioaktive Probe abgeschieden und anschließend getrocknet wird.
In einem dritten Raum wird mit einem Zählrohr die Aktivität dieser Probe gemessen. Die Zählrohre werden in der Institutswerkstatt aus Aluminiumrohren von ca. 2 cm Durchmesser und 6 cm Länge hergestellt. Deren Wandstärke wird zuvor durch Abdrehen auf der Drehbank auf ca. 0,1 mm verringert, damit die zu messende β-Strahlung nicht bereits in der Zählrohrwand absorbiert wird. Ein mittig eingeführter Stahldraht wird mithilfe von beidseitig angebrachten Hartgummistopfen gegen das Rohr isoliert, ein ebenfalls eingebettetes Glasröhrchen dient zum Auspumpen und Füllen des Zählrohrs mit einem gasförmigen Argon-Alkohol-Gemisch.
Bei der Messung wird das Zählrohr zur Abschirmung äußerer Einflüsse waagrecht in einen zerlegbaren Bleiblock eingebettet, in dem sich unterhalb des Zählrohres eine Öffnung befindet, in die ein Blei-„Schiffchen“ eingeschoben werden kann, in dessen Mulde zuvor das Filter mit der Probe eingeklebt worden ist. Auf diese Art liegt die radioaktive Probe weitgehend formgerecht an der Außenwand des Zählrohrs an. Zur Spannungsversorgung des Zählrohrs (1000–1500 Volt Gleichspannung) dienen mehrere hintereinandergeschaltete Anodenbatterien, über einen hochohmigen Widerstand ist der Zähldraht an den positiven, das Gehäuse an den negativen Pol der Batterie angeschlossen.
Gelangt ein von der zu messenden Probe emittiertes β−-Teilchen (Elektron) durch die Aluminiumwand des Zählrohrs in das Rohrinnere, so wird es im elektrischen Feld zum Zähldraht hin beschleunigt. Auf diesem Weg werden die von ihm getroffenen Argonatome ionisiert, die hierbei gebildeten Elektronen fliegen ebenfalls zum Zähldraht und bilden unterwegs durch Ionisation weitere Elektronen. Diese Elektronenlawine fließt über Zähldraht und Widerstand zum geerdeten positiven Pol ab. Durch einen Röhrenverstärker wird dieser Stromimpuls verstärkt und von einem mechanischen Zählwerk registriert. Die Alkoholfüllung des Zählrohrs bewirkt ein selbsttätiges Erlöschen der Entladung nach kurzer Zeit, so dass das Zählrohr dann zum Nachweis eines weiteren Elektrons bereit ist.
Das Arbeiten mit unwägbaren radioaktiven Substanzmengen
Bei der Suche nach den Transuranen stehen die Forscher am KWI vor dem Problem, in einer relativ großen Menge einer wasser- oder säurelöslichen Uranverbindung (ca. 10 bis 100 g) eine durch die Neutronenbestrahlung in kleinsten, unwägbaren Mengen gebildete neue, radioaktive Substanz – wesentlich weniger als 1 Pikogramm (10−12 Gramm) – chemisch zu identifizieren.
Auch für die empfindlichsten damals bekannten Analysenverfahren, die Spektralanalyse und die Massenspektrometrie, ist diese Menge jedoch viel zu gering, damit geht es also nicht. Der Nachweis der radioaktiven Strahlung dieser neuen Substanz ist jedoch weitaus empfindlicher als die beiden ebengenannten Analysenverfahren. Für das Arbeiten mit unwägbaren radioaktiven Substanzen hat man deshalb seit der Entdeckung des Radiums (1898) spezielle „radiochemische“ Arbeits- und Analysentechniken entwickelt, in denen diese Nachweisstärke mit der Selektivität chemischer Trennungen kombiniert wird. Hierbei werden immer wieder die beiden nachfolgenden chemischen Trennmethoden angewandt.
Die Abtrennung durch Mitfällung
Es wurde vermutet, dass es sich bei einem oder mehreren der von Fermi beobachteten fünf Aktivitäten um das unbekannte Element 93 handelt. Ein Blick in das Periodensystem der chemischen Elemente in seiner damaligen Form zeigt das Element 93 unterhalb von Rhenium, es sollte also zu ihm und den darüber stehenden Elementen Mangan und Masurium homolog sein und seine chemischen Verbindungen müssten sich ähnlich wie die von Rhenium, Masurium und Mangan verhalten. Masurium konnte allerdings nicht zum Vergleich herangezogen werden, da keine Verbindungen dieses Elements bekannt sind. Es bleiben für eine Ähnlichkeitsbetrachtung also nur Rhenium und Mangan.
Gesucht wird nun ein chemisches Reagens, mit dem aus einer wässrigen Lösung von Rhenium und Uran das Rhenium ausgefällt werden kann, Uran jedoch in Lösung bleibt. Ein solches Reagens ist Schwefelwasserstoff, er bildet mit den ebengenannten Elementen Sulfide, jedoch mit folgenden unterschiedlichen Eigenschaften.
Uransulfid: löslich in verdünnten Säuren Mangansulfid: löslich in verdünnten Säuren Rheniumsulfid: unlöslich sogar in starker (unverdünnter) Salzsäure Aus der unterschiedlichen Löslichkeit von Mangan- und Rheniumsulfid kann man schließen, dass ein Sulfid des Elements 93 mindestens ebenso unlöslich in Salzsäure ist wie Rheniumsulfid.
Man löst also das bestrahlte Uran in starker Salzsäure, gibt eine kleine Menge (einige Milligramm) eines löslichen Rheniumsalzes dazu und leitet gasförmigen Schwefelwasserstoff ein. Es bildet sich unlösliches schwarzes Rheniumsulfid Re2S7, wobei das Sulfid des neu gebildeten Elements mitgerissen werden sollte, falls es sich um das Element 93 handelt.
Man bezeichnet diesen Vorgang als eine Mitfällung. Die äußerst geringen Substanzmengen eines chemisch verwandten Elements werden ebenfalls in Form ihres Sulfids in das Kristallgitter des Rheniumsulfids eingebaut und mit ausgefällt. Man filtriert den Niederschlag ab und hat auf diese Weise die durch die Bestrahlung entstandenen Isotope des erwarteten Elements 93 vom Uran abgetrennt.
Wird damit gerechnet, dass sich unter den Reaktionsprodukten des Urans auch die Elemente 94 bis 96 befinden, so empfiehlt sich ebenfalls eine Mitfällung als Sulfid, diesmal jedoch in mittelstarker Salzsäure und mit einem Platinsalz als Trägersubstanz; das Element 93 wird hierbei mitgefällt.
Die Trennung homologer Substanzgemische durch fraktionierte Kristallisation
Soll die aktive Substanz von dem inaktiven Träger getrennt werden, so erweist sich die bei der Mitfällung erwünschte Eigenschaft einer chemischen Ähnlichkeit der Verbindungen nun, bei der Trennung, als großes Hindernis. Hilfe bringt hier das Verfahren einer fraktionierten Kristallisation (oder fraktionierten Fällung), das um 1885 von Carl Auer von Welsbach entwickelt wurde, um die chemisch sehr ähnlichen Elemente der Lanthaniden (Seltene Erden; Elemente 57 bis 71) voneinander zu trennen.
Für die Radiochemie erprobt wurde dieses Verfahren von Marie Curie in den Jahren 1899–1902 bei der Reindarstellung des Radiums aus den Verarbeitungsrückständen der Pechblende.[2]
Sie stand vor der Aufgabe, von dem aus den Rückständen bereits isolierten radiumhaltigen Bariumchlorid (ca. 8 kg BaCl2 pro Tonne Verarbeitungsrückstände) das Radium in wägbaren Mengen vom Barium abzutrennen, um es spektralanalytisch untersuchen und sein Atomgewicht bestimmen zu können.
Sie löste hierzu das Bariumchlorid (in Mengen von etwa 1 kg) in heißem destillierten Wasser und kochte die Lösung so lange ein, bis sich erste Kristalle zeigten. Beim Abkühlen kristallisierte dann ein Teil des Bariumchlorids aus, es bildeten sich am Boden der Schale schöne, festhaftende Kristalle (Fraktion A; Kopffraktion), von denen die überstehende Mutterlauge nach dem Erkalten leicht abgegossen werden konnte. Die Mutterlauge wurde dann in einer zweiten (kleineren) Schale wieder bis zur Sättigung eingedampft; nach dem Abkühlen und „Dekantieren“ (Abgießen der Mutterlauge) erhielt sie die Kristallfraktion B (Schwanzfraktion). Beim Vergleich der Aktivität beider Kristallfraktionen stellte M. Curie fest, dass die Fraktion A ungefähr fünfmal stärker radioaktiv war als Fraktion B. Der Grund hierfür ist die geringere Wasserlöslichkeit von Radiumchlorid gegenüber Bariumchlorid, es wurde deshalb (obwohl es in nur unwägbar kleinsten Mengen in der Lösung vorhanden war) in der ersten Kristallfraktion des Bariumchlorids bei der Mitfällung „angereichert“.
Mme Curie musste diesen Vorgang (Lösen, Eindampfen, Auskristallisieren, Dekantieren) unzählige Male und an immer wieder neuen Mengen von radiumhaltigem Bariumchlorid wiederholen, um schließlich einige Milligramm bariumfreies Radium zu erhalten. Eine ausführliche Beschreibung dieser sehr mühevollen und äußerst langwierigen Arbeiten bleibt einem Artikel über die Entdeckung des Radiums vorbehalten. Im Zusammenhang mit der Kernspaltung sind jedoch noch folgende Hinweise von M. Curie interessant:
Verwendet man zur Lösung des Barium-Radium-Chlorids anstatt Wasser verdünnte oder gar starke Salzsäure, so wird die Löslichkeit beider Chloride verringert und der Trenneffekt zwischen beiden Komponenten außerdem beträchtlich vergrößert; die Anreicherung des Radiums in der Kopffraktion ist also erheblich größer als bei einer wässrigen Lösung. Noch größer ist die Anreicherung des Radiums in der Kopffraktion, wenn die Isolierung des radiumhaltigen Bariums aus den Pechblenderückständen nicht mit Barium- und Radiumchlorid, sondern in Form ihrer Bromide (also mit Bariumbromid + Radiumbromid) erfolgt.
Die Messung der Aktivität mit dem Zählrohr
Die ausgefällte inaktive Trägersubstanz, in deren Kristallgitter die nahezu gewichtslose Menge des radioaktiven Reaktionsproduktes eingeschlossen ist, wird abfiltriert und getrocknet, das Filter in ein Bleischiffchen eingeklebt und in den Bleiblock des Zählrohrs eingeschoben. Die β−-Strahlung des Präparates wird dann in bestimmten Zeitabständen gemessen. Die Dauer einer Zählrohrmessung kann frei gewählt werden, naturgemäß muss sie um so länger sein, je schwächer die Aktivität der Probe ist. Errechnet wird aus dem Messwert dann die Anzahl der Zählimpulse pro Minute.
Vor oder nach jeder Messung eines radioaktiven Präparates muss der Nulleffekt des Zählrohrs gemessen und dessen Wert von der Aktivität des Präparats abgezogen werden. Man versteht unter dem Nulleffekt die Anzahl der Zählimpulse pro Minute, die vom Zählrohr registriert werden, wenn sich keine radioaktive Probe im Bleiblock befindet. Ursache für diesen Leerwert von etwa 10–20 Impulsen/Minute ist im Wesentlichen die Höhenstrahlung.
Trägt man dann die jeweils ermittelte Aktivität der Probe in Abhängigkeit von der Zeit graphisch auf, so lässt sich aus dem Kurvenverlauf die Halbwertszeit des Strahlers bestimmen. Enthält der Niederschlag mehrere radioaktive Isotope, so lässt sich auch hier die HWZ für jedes Isotop ermitteln, wenn diese Werte nicht zu nahe beieinander liegen.
Ein Transuran unter den Reaktionsprodukten
Bis zum Sommer 1938 verläuft die Arbeit am KWI in Berlin unbeeinflusst von den politischen Ereignissen in Deutschland. Die Kernphysikerin Meitner, der Radiochemiker Hahn und der Analytiker Straßmann (der Mann für die Trennversuche) ergänzen sich auf das Beste. Das Team glaubt herausgefunden zu haben, dass es sich bei den erfolgreich abgetrennten Substanzen um drei betastrahlende Uranisotope sowie sieben Folgeprodukte handelt, die sich bei bzw. nach der Neutronenbestrahlung von Uran gebildet haben.
Uran Element 93 Element 94 Element 95 Element 96 Element 97 10 sec −> 2,2 min −> 59 min −> 66 h −> 2,5 h −> ? 40 sec −> 16 min −> 5,7 h −> 60 d −> ? 23 min −> ? Alle diese radioaktiven Körper können sowohl durch schnelle als auch, mit viel besserer Ausbeute, durch thermische Neutronen erzeugt werden.
Für die an dritter Stelle aufgeführte Substanz (mit der HWZ von 23 min) können allerdings keine Folgeprodukte gefunden werden, man vermutet deshalb, dass es sich bei seinem ersten Folgeprodukt, also Element 93, um ein sehr langlebiges Isotop handelt, das während der Beobachtungsdauer nicht merklich unter Strahlenemission zerfällt. Wie später durch McMillan nachgewiesen wurde, handelte es sich diesem einen Falle tatsächlich nicht um ein Spaltprodukt von U-235, sondern um eine Reaktion des Neutrons mit Uran-238 nach der ursprünglich angenommenen Reaktion:
Damit war wirklich ein Transuran entstanden.
Zur Abtrennung der Uranisotope werden zunächst die Elemente 93 bis 96 durch Fällung an Platinsulfid entfernt und dann wird im Filtrat das zur Bestrahlung eingesetzte Uran als Natrium-Magnesium-Uranylacetat ausgefällt. Wenn diese beiden Fällungen sehr schnell erfolgen, kann im Uranniederschlag der 40s-Körper noch gemessen werden.
In zwei Veröffentlichungen[3][4] des Jahres 1937 werden die gewonnenen Erkenntnisse über die chemischen und kernphysikalischen Eigenschaften der Transurane zusammengefasst. Die chemische Ähnlichkeit mit den homologen Elementen Rhenium, Osmium, Iridium und Platin und damit auch die Einordnung in das periodische System als Transurane erscheint gesichert.
Die kernphysikalischen Befunde stützen diese Interpretation, wenn es auch Probleme macht, zu verstehen, wie aus Uran-238 drei verschiedene Produktlinien entstehen können. An eine Namensgebung für die neuen chemischen Elemente denkt das Berliner Team jedoch nicht.
Im Juli 1938 erscheint dann von Hahn, Meitner und Straßmann die letzte gemeinsame Veröffentlichung.[5]
Lise Meitner war als Österreicherin von den Rassegesetzen Hitlers anfangs nicht betroffen. Mit dem Anschluss Österreichs an Deutschland im März 1938 änderte sich jedoch diese Situation, sie emigrierte deshalb Mitte Juli 1938 nach Stockholm an das Nobel-Institut für Physik. Mit Otto Hahn stand sie jedoch in regem Briefwechsel; sie war so über alle weiteren Arbeiten am KWI informiert.
Die Neutronenbestrahlung von Uran in Paris 1937–1938: Die 3,5-Stunden-Aktivität
Von I. Curie und P. Savitch in Paris war 1937 beim Bestrahlen von Uran mit Neutronen ein β−-Strahler mit einer Halbwertszeit von 3,5 Stunden gefunden worden, dessen chemische Identifizierung sich jedoch als außerordentlich schwierig erwies. Da sich die Substanz zusammen mit Lanthan, einem Element aus der 3. Gruppe des periodischen Systems, ausfällen lässt, nehmen Curie und Savitch anfangs an, dass es sich um das nächsthöhere Element dieser Gruppe, also Actinium handelt. Nur wenig später berichten sie jedoch, dass der 3,5 h-Körper zwar ganz ähnliche Eigenschaften wie Lanthan hat, sich von Actinium und Lanthan jedoch durch fraktionierte Fällung trennen lässt, es sich also nicht um Actinium oder Lanthan handeln kann. Sie vermuten deshalb (Juli 1938), dass der 3,5 h-Körper ein Transuran sein müsse. Ungeachtet seiner sehr großen Ähnlichkeit mit Lanthan können sie sich jedoch vorstellen, „dass dieser Körper die Kernladungszahl 93 hat und es sich bei den von Hahn, Meitner und Straßmann bisher gefundenen Transuranen um die Elemente 94 bis 97 handelt.“[6]
Der chemische Nachweis einer Kernspaltung des Urans
Berlin, Herbst 1938: Die „Radium“-Isotope
Mitte Oktober 1938 wird diese Veröffentlichung[6] in Berlin bekannt. Das waren neue Gesichtspunkte und zugleich eine Herausforderung für Hahn und Straßmann. Das Berliner Team hatte die Abtrennung der Reaktionsprodukte stets mit einer gemeinsamen Fällung an Platinsulfid begonnen. Dabei fällt der 3,5 h-Körper, wie Curie und Savitch berichten, nicht mit. Deren Trennungsgang beginnt mit einer Fällung an schwerlöslichem Kalium-Lanthan-Sulfat.
Hahn und Straßmann gehen bei der Überprüfung dieser Ergebnisse von folgenden Überlegungen aus. Actinium, das zumindest für einen gewissen Zeitraum von Paris postuliert worden war, könnte aus Uran in folgenden Schritten entstehen:
Das auf den Urankern auftreffende Neutron schlägt in einem (n, α)-Prozess ein α-Teilchen aus dem Kern heraus und es entsteht ein Thoriumisotop:
Aus dem Thorium-235 entsteht durch α-Zerfall Radium
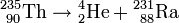 oder in abgekürzter Schreibweise
oder in abgekürzter Schreibweise 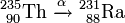
und aus diesem durch β−-Zerfall Actinium:
Bei einer Mitfällung des so entstehenden Actiniums an Kalium-Lanthan-Sulfat kann dann aber das intermediär gebildete Radium-231 als schwerlösliches Radiumsulfat mitgerissen werden und so das Actinium in den Niederschlag begleiten. Bei der anschließenden Fraktionierung würde Radium in die Kopffraktion gelangen und so das Ergebnis verfälschen.
Hahn und Straßmann prüfen diese Hypothese, indem sie aus der bestrahlten Probe Bariumchlorid ausfällen. Diese Fällung ist tatsächlich radioaktiv, es muss also Radium mitgefällt worden sein. Aufgrund der gemessenen unterschiedlichen Halbwertszeiten enthält die Fällung sogar mindestens drei betastrahlende Radiumisotope, die ihrerseits drei betastrahlende Actiniumisotope nachbilden und diese wiederum drei nicht näher charakterisierte Thoriumisotope.
In einer Anfang November 1938 erscheinenden Mitteilung[7] stellen sie dies so dar:
![{Ra}^{}_{1} \ \xrightarrow[\mathrm{\sim} \ \text {25 Min.}]{\text{ }\mathrm{\beta}} \mathrm {\ Ac}^{}_{1} \ \xrightarrow[\mathrm{\sim} \ \text{40 Min.}]{\text{ }\mathrm{\beta}} \mathrm {\ Th?}](/pictures/dewiki/54/66b936be79c5ced93aab50f8ae203367.png)
![{Ra}^{}_{2} \ \xrightarrow[\mathrm{\sim} \ \text {110 Min.}]{\text{ }\mathrm{\beta}} \mathrm {\ Ac}^{}_{2} \ \xrightarrow[\mathrm{\sim} \ \text{4 Stdn.}]{\text{ }\mathrm{\beta}} \mathrm {\ Th?}](/pictures/dewiki/51/3c33e8ab2e56f10d7e6abc0db0fb6a8e.png)
![{Ra}^{}_{3} \ \xrightarrow[\text{mehrere Tage}]{\text{ }\mathrm{\beta}} \mathrm {\ Ac}^{}_{3} \ \xrightarrow[\mathrm{\sim} \ \text{60 Std.}]{\text{ }\mathrm{\beta}} \mathrm {\ Th?}](/pictures/dewiki/54/61b0e974acdd49b1ca098927818f021d.png)
Dieser Befund ist jedoch in mehrfacher Hinsicht problematisch. Gefunden werden drei Radium-Actinium-Paare, die sich in ihren Halbwertszeiten unterscheiden. Sie müssen alle die gleiche Massenzahl 231 haben, weil der von einem Neutron getroffene Atomkern Uran-238 sich nur nach folgendem, bereits in seinen Einzelschritten beschriebenen Schema in Radium umgewandelt haben kann:
Von diesem Radium-231 gibt es aber offensichtlich die drei „Versionen“ Ra1, Ra2 und Ra3 mit verschiedenen Halbwertszeiten!
Nun waren durchaus Fälle bekannt, bei denen es zwei Versionen eines Atomkerns mit verschiedenen Halbwertszeiten gibt; man nennt dieses Phänomen Kernisomerie und führt es auf verschiedene Energiezustände desselben Kerns zurück. Eine dreifache Kernisomerie, die noch dazu beim radioaktiven Zerfall auch auf die Folgeprodukte Actinium-231 übertragen wird, war jedoch gänzlich neu und schwer zu erklären.
Aber auch der postulierte (n, α)-Prozess macht Probleme, weil er schon mit langsamen Neutronen ablaufen müsste, zum Herausschlagen eines α-Teilchens aus dem getroffenen Urankern aber eigentlich energiereiche (schnelle) Neutronen erforderlich wären. Andererseits war das gleiche Phänomen aber auch bereits bei der Bestrahlung von Thorium beobachtet worden, wo drei Radiumisotope mit unterschiedlichen Halbwertszeiten (<1 Minute, 15 Minuten, 4 Stunden) dem Radium-229 zugeordnet wurden[8]:
Über diesen Stand der Erkenntnisse berichtet Otto Hahn Mitte November 1938 in Kopenhagen am Bohrschen Institut. Er trifft dort auch Lise Meitner und Otto Frisch. Die Kernphysiker Meitner, Bohr und Frisch sind skeptisch hinsichtlich der Kernisomerie, haben aber für die Befunde auch keine befriedigende Erklärung. Weitergehende Experimente sind also notwendig; es geht um die Frage: „Sind die mit Ra1, Ra2 und Ra3 bezeichneten Radiumisotope wirklich Radium?“
Um dies zu entscheiden, muss man die in der Barium-Radium-Fraktion in nur unwägbaren Mengen vorhandenen radioaktiven Ra-Isotope vom Barium abtrennen oder zumindest anreichern. Möglich ist dies durch die bereits beschriebene fraktionierte Kristallisation von Bariumchlorid oder Bariumbromid.
Ehe es jedoch hierzu kommt, wird noch ein viertes, sehr kurzlebiges Radiumisotop 231Ra gefunden, Hahn und Straßmann ändern deshalb die Probenbezeichnungen der bisherigen drei Zerfallsreihen. Die Halbwertszeiten sind mittlerweile auch genauer bestimmt, es ergibt sich deshalb folgendes Schema:
![{Ra \ I?}^{}_{} \ \xrightarrow[\mathrm{&amp;lt;} \ \text {1 Min.}]{\text{ }\mathrm{\beta}} \mathrm {\ Ac \ I \ } \xrightarrow[\mathrm{&amp;lt;} \ \text{30 Min.}]{\text{ }\mathrm{\beta}} \mathrm {\ Th?}](/pictures/dewiki/101/e4f81d4906f37b5203f29a9e367e4e44.png)
![{Ra \ II}^{}_{} \ \xrightarrow[\text {14}\ \mathrm{\pm} \ {2 Min.}]{\text{ }\mathrm{\beta}} \mathrm {\ Ac \ II \ } \xrightarrow[\mathrm{\sim} \ \text{2,5 Std.}]{\text{ }\mathrm{\beta}} \mathrm {\ Th?}](/pictures/dewiki/50/247b6fc55408b330f3deaff441463e7e.png)
![{Ra \ III}^{}_{} \ \xrightarrow[\text {86}\ \mathrm{\pm} \ {6 Min.}]{\text{ }\mathrm{\beta}} \mathrm {\ Ac \ III \ } \xrightarrow[\mathrm{\sim}\ \text{mehrere Tage?}]{\text{ }\mathrm{\beta}} \mathrm {\ Th?}](/pictures/dewiki/49/1f9e1800a9c004578b228ca91b60d4be.png)
![{Ra \ IV}^{}_{} \ \xrightarrow[\text {250-300 Std.}]{\text{ }\mathrm{\beta}} \mathrm {\ Ac \ IV \ } \xrightarrow[\text{40 Std.}]{\text{ }\mathrm{\beta}} \mathrm {\ Th?}](/pictures/dewiki/56/83389117fcbd083a156e2581d78f3aba.png)
Trennversuche
Nur dem Umstand, dass die Laborjournale von Fritz Straßmann zum großen Teil erhalten sind, ist es zu verdanken, dass Einzelheiten über Versuchsdurchführung und Messergebnisse des KWI der letzten Wochen des Jahres 1938 auch heute noch allgemein bekannt und zugänglich sind.
Die Originale befinden sich im Nachlass von Fritz Straßmann im Landeshauptarchiv Koblenz, das Protokollheft „Chem. II“ (Nr. 192, November 1938 bis Februar 1939) jedoch im Deutschen Museum. Faksimiles einzelner Protokollseiten sind in dem im Abschnitt „Sonstige Literatur“ erwähnten Buch von F. Krafft, dem Band 95 der Landesarchivverwaltung Rheinland-Pfalz sowie den beiden Aufsätzen von G. Herrmann zu finden.
Die in den folgenden Versuchen genannten Details (Probemengen, Bestrahlungsdauer, Messdatum, Messergebnisse etc.) sind diesen Laborjournalen entnommen.
Die Fraktionierung mit Bariumchlorid
Die erste Radium-Barium-Fraktionierung wird am 25. November 1938 durchgeführt. Sie sei exemplarisch eingehender beschrieben:
11 g Ammoniumdiuranat waren 16 Stunden mit verlangsamten (thermischen) Neutronen bestrahlt worden, die Bestrahlung wird um 11:36 Uhr beendet. Die Probe wird dann in Salzsäure gelöst, und nach Zugabe von 1,5 g Bariumchlorid (als Trägersubstanz) wird das Barium mit starker Salzsäure ausgefällt; die durch die Bestrahlung gebildeten Radiumisotope werden hierbei mitgefällt. Der Niederschlag wird abfiltriert und so von der Uranlösung getrennt. Das Gemisch von Barium- und Radiumchlorid wird dann in heißem Wasser gelöst und so lange tropfenweise mit starker Salzsäure versetzt, bis die Lösung trüb wird. Durch Abkühlen der Lösung kristallisiert ein Teil des Bariumchlorids aus und wird abfiltriert; durch weiteres Abkühlen des Filtrats werden zwei weitere Kristallfraktionen gewonnen. Nach der Abtrennung der ersten Kristallfraktion kann dann bereits mit den Zählrohrmessungen begonnen werden. In der Zeit von 12:05 Uhr bis 13:22 Uhr wird dann die Zählrate jeweils gleicher Mengen der drei Fraktionen (350 mg BaCl2) alternierend mit dem gleichen Zählrohr gemessen. Die Aktivität der Proben nimmt während dieser Zeitspanne kontinuierlich ab (für die erste Fraktion von 387 auf 132 Impulse/Minute), dies ist durch den radioaktiven Zerfall von RaII und RaIII bedingt. Eine sprunghafte Abnahme der Aktivität zwischen der ersten und zweiten oder der zweiten und dritten Kristallfraktion ist jedoch nicht zu erkennen. Eine Anreicherung der Radiumisotope in der Kopffraktion hat also nicht stattgefunden.
Die Fraktionierungen mit Bariumbromid
Nur drei Tage später wird der nächste Versuch durchgeführt. Die Uranprobe ist diesmal 45 Stunden lang bestrahlt worden, die Mitfällung der Reaktionsprodukte erfolgt wiederum an Bariumchlorid, die anschließende Fraktionierung jedoch mit Bariumbromid. Hierzu wird die Mischung aus Barium- und Radiumchlorid in Form ihrer Carbonate ausgefällt und die Fällung in Bromwasserstoffsäure HBr gelöst. Durch tropfenweise Zugabe weiterer HBr wird die Lösung fraktioniert kristallisiert. Vier Kristallfraktionen von Bariumbromid sind diesmal das Ergebnis.
Mit dem Beginn der Messungen wartet man etwa eine Stunde, dann liegt nur noch RaIII vor. Drei dieser vier Fraktionen werden abwechselnd gemessen. Auch hier ist die Aktivität bei allen Proben zum gleichen Zeitpunkt die gleiche, sie nimmt der HWZ des RaIII entsprechend stetig ab.
Bei einem dritten Versuch, am 8. Dezember, mit lang bestrahltem Uran, erfolgt eine Fraktionierung mit dem langlebigen RaIV, wiederum mit Bromid. Auch hier das gleiche Ergebnis, alle Fraktionen haben die gleiche Aktivität, eine Anreicherung des RaIV findet ebenfalls nicht statt.
Kontrollversuche
Bei der Suche nach einer plausiblen Erklärung für dieses anomale Verhalten des Radiums halten Hahn und Straßmann es für möglich, dass die Radiumisotope nur deshalb nicht in der Kopffraktion der Bariumsalze angereichert werden, weil ihre Konzentration in den Trägersubstanzen zu gering ist; jedenfalls wesentlich geringer als bei den klassischen Arbeiten zur Isolierung des Radiums.
Zur Gegenkontrolle versetzen sie deshalb Bariumchlorid mit unterschiedlichen Mengen an natürlich radioaktiven Radiumisotopen (224Ra bzw. 228Ra; vgl. Indikatorversuche) und fraktionierten. Diese (wirklichen) Radiumisotope reicherten sich, auch wenn sie in nur sehr geringen Konzentrationen dem Barium beigemischt sind, erwartungsgemäß in der Kopffraktion des Bariums an.
Indikatorversuche
Die bisher beschriebenen Versuche haben also übereinstimmend gezeigt, dass bei der fraktionierten Kristallisation der Radiumisotope RaII, RaIII und RaIV mit Bariumchlorid oder Bariumbromid nicht die für Radium zu erwartende Anreicherung in der Kopffraktion erfolgt. Die natürlichen Radiumisotope 224Ra und 228Ra hatten sich hingegen, in getrennten Versuchsreihen, wie gewohnt verhalten.
Nun lag es nahe, die beiden Versuchsreihen zu vereinen: das aus dem Uran durch Neutronenbestrahlung entstandene „künstliche“ Radium mit natürlichem Radium zu vermischen und gemeinsam zu fraktionieren. Das natürliche Radium ist dann der Indikator für das chemische Verhalten dieses Elements unter den gewählten Bedingungen; ein künstliches Radium muss sich genau so verhalten.
Hahn und Straßmann betrachten einen solchen Versuch als experimentum crucis. Sie verwenden als Indikator das Radiumisotop Mesothorium 1 (MsTh1; 228Ra), einen β−-Strahler, der 1908 von Hahn entdeckt worden war. Er ist die Tochtersubstanz des natürlichen Thoriums, des Anfangsgliedes der Thorium-Zerfallsreihe:
![{}^{232}_{\ 90} \mathrm {Th}\ \xrightarrow[{1,4} \ \cdot \ {10^1}{^0} {\ a}]{\text{ }\mathrm{\alpha}} {}^{228}_{\ 88} \mathrm {Ra\ (MsTh1)} \ \xrightarrow[{6,7\ a}]{\text{ }\mathrm{\beta}\mathrm{^-}} \ {}^{228}_{\ 89} \mathrm {Ac\ (MsTh2)} \ \xrightarrow[{6,13\ h}]{\text{ }\mathrm{\beta}\mathrm{^-}} \ {}^{228}_{\ 90} \mathrm {Th} \ \xrightarrow[{1,9\ a}]{\text{ }\mathrm{\alpha}} \](/pictures/dewiki/97/a376abb58717cddf31e42850d276cb01.png) ……
……
Aus dem Radium-228 geht das Actiniumisotop Mesothorium2 (MsTh2; Actinium-228) hervor, ebenfalls eine frühe Entdeckung Hahns. Es dient als Indikator für ein gleichartiges Experiment mit dem künstlichen Actinium (vgl. Abschnitt AcIV - MsTh2).
RaIII - MsTh1
Der Versuch zur Prüfung von RaIII wird am 17. Dezember 1938 durchgeführt. Nachdem eine Uranprobe über Nacht bestrahlt worden war, bleibt sie 2,5 Stunden stehen, um das kurzlebige RaI und RaII zerfallen zu lassen. Die Folgeprodukte AcI und AcII werden dann entfernt und als Indikator wird gereinigtes MsTh1 zugegeben. Zur Mitfällung des RaIII und des MsTh1 werden 3 Gramm Bariumbromid zugesetzt.
Die Fraktionierung ergibt drei Fraktionen mit je 500 mg BaBr2. Um zu entscheiden, wie sich die beiden Radium-Isotope verhalten, muss die Aktivität der Fraktionen über mehrere Tage hinweg gemessen werden, da es hierzu ja drei Beiträge gibt:
RaIII, das mit einer HWZ von 86 Minuten zerfällt, MsTh1 (228Ra), dessen Aktivität während der Versuchsdauer konstant bleibt (HWZ: 6,7 a), MsTh2 (228Ac), das bei der Fraktionierung in der Mutterlauge zurückbleibt, aber während der Messdauer aus MsTh1 nachgebildet wird (HWZ: 6,13 h). Erst anhand einer graphischen Auswertung kann dann das Ergebnis ermittelt werden. Die Kurven zeigen in den ersten Stunden einen steilen Aktivitätsabfall, der durch den Zerfall von RaIII verursacht ist. Dieser Abfall wird gebremst und schließlich in einen Aktivitätsanstieg umgemünzt durch das aus MsTh1 nachgebildete MsTh2, das nach ca. 60 Stunden seinen Endwert erreicht hat und dann im radioaktiven Gleichgewicht mit seiner langlebigen Muttersubstanz MsTh1 ist. Dieser Endwert liegt für die drei Fraktionen bei 67,6 / 25 / 11 Impulsen/Minute. Er besagt, dass das MsTh1 in den Bariumbromidfraktionen 1 bis 3 in Konzentrationen vorhanden ist, die sich wie 6,1 : 2,3 : 1 verhalten; das natürliche Radiumisotop hat sich also wie erwartet in der Kopffraktion stark angereichert.
Der Aktivitätsanteil des RaIII bei jeder der drei Fraktionen ergibt sich, wenn von der jeweils gemessenen Aktivität die Anteile abgezogen werden, die dem MsTh1 und dem nachgebildeten MsTh2 zuzuordnen sind. Trägt man diese bereinigten Messwerte in Abhängigkeit von der Zeit in ein Diagramm mit halblogarithmischem Maßstab ein, so liegen die Messpunkte für jede Fraktion auf einer Geraden, deren Neigung der HWZ des RaIII entspricht; extrapoliert man diese Geraden auf den Zeitpunkt Null, so ergeben sich für die drei Fraktionen Aktivitäten von 81, 72 und 81 Impulsen/Minute. Innerhalb der Fehlergrenzen der Messungen (±10 % angesichts der geringen Zählraten) ist die Konzentration des RaIII in den drei Bariumbromidfraktionen also konstant, eine Anreicherung hat nicht stattgefunden. RaIII verhält sich also auch bei diesem Versuch nicht wie der zugesetzte Radiumindikator MsTh1, sondern wie natürliches Barium.
AcIV - MsTh2
Wenn die Radiumisotope kein Radium sind, sondern Barium, dann wird aus der obengenannten Zerfallsreihe mit den vermeintlichen Radiumisotopen (vgl. Berlin, Herbst 1938: Die „Radium“-Isotope)
-
- Ra → Ac → Th → ?
die neue Reihe
-
- Ba → La → Ce → ?
Das Ac-Isotop ist also kein Actinium, sondern sollte Lanthan sein. Auch diese Folgerung prüfen Hahn und Straßmann in einem weiteren Indikatorversuch zwei Tage später nach. Sie unterziehen bei einer mehrere Tage lang bestrahlten Uranprobe das aus RaIV nachgebildete AcIV (HWZ: 40 Stdn) einer fraktionierten Kristallisation mithilfe von Lanthanoxalat. Die Oxalate von Lanthan und Actinium sind in verdünnten Säuren schwer löslich, Actinium-Oxalat jedoch nicht so sehr wie Lanthan-Oxalat; bei einer fraktionierten Kristallisation müsste sich also Actinium-Oxalat in der letzten Fraktion, der Schwanzfraktion, anreichern.
Als Indikator wird das natürlich radioaktive Ac-Isotop MsTh2 (HWZ: 6,1 Stdn) zugeben. MsTh2 reichert sich, wie erwartet, in der Schwanzfraktion an, das AcIV jedoch ist gleichmäßig über alle vier Fraktionen verteilt; es ist also kein Actinium, es verhält sich wie Lanthan.
Berlin, Dezember 1938: Der chemische Nachweis von Barium
Alle diese Trenn-, Kontroll- und Indikatorversuche zeigen also, dass die Körper RaII, RaIII und RaIV, die man bisher für Radiumisotope gehalten hatte, keine Isotope des chemischen Elements Radium sein können. Sie verhalten sich chemisch genau so wie das inaktive Trägerelement Barium. Hieraus müssen Hahn und Straßmann den einzig möglichen Schluss ziehen: Die vermeintlichen Radiumisotope sind kein Radium, es sind radioaktive Isotope des chemischen Elements Barium.
Die Ergebnisse dieser Untersuchungen werden von Hahn und Straßmann in einem Bericht zusammengefasst, der am 22. Dezember 1938 bei der Redaktion der Zeitschrift „Die Naturwissenschaften“ eingereicht wird und bereits am 6. Januar 1939 erscheint[9]. Die Autoren geben ihre „seltsamen Ergebnisse nur zögernd“ bekannt. Als Chemiker zweifeln sie nicht daran, dass es sich bei den vermeintlichen Radiumisotopen um radioaktive Bariumisotope handelt. Als der Physik „in gewisser Weise nahestehende Kernchemiker“ halten sie sich jedoch mit einer physikalischen Deutung des Vorgangs zurück, da er allen bisherigen Erfahrungen der Kernphysik widerspricht. In einem Brief an Lise Meitner vom 19. Dezember 1938 wird Hahn jedoch deutlicher, indem er schreibt: „Vielleicht kannst Du irgend eine phantastische Erklärung vorschlagen. Wir wissen dabei selbst, dass es (Anmerkung: der Urankern) eigentlich nicht in Ba zerplatzen kann.“
Indikatorversuch mit Radiumisotop ThX
In einer weiteren Arbeit[10] die Ende Januar 1939 eingereicht wird, veröffentlichen Hahn und Straßmann dann die Messkurven ihres Indikatorversuchs mit RaIII-MsTh1 und Bariumbromid und sie berichten über die Ergebnisse weiterer Indikatorversuche mit RaIII und RaIV (wobei sie an Stelle von RaIII und RaIV nun BaIII und BaIV schreiben). Der Indikator ist das Isotop 224Ra (Thorium X), ein α-Strahler, der über ein β−-strahlendes Folgeprodukt nachgewiesen wird. Die Fraktionierung des Radium-Barium-Gemisches erfolgt durch Fällung der schwerlöslichen Chromate, die sich besonders gut für diesen Zweck eignen. Die Auswertung der Messkurven bringt wiederum das gleiche Ergebnis: das „echte“ Radiumisotop (ThX) wird in der Kopffraktion von BaCrO4 sehr stark angereichert, das vermeintliche Radium (BaIII bzw. BaIV) ist gleichmäßig auf die einzelnen BaCrO4-Fraktionen verteilt.
Kreislaufversuch
In einem letzten Indikatorexperiment prüfen Hahn und Straßmann, ob inaktives Barium und die vermeintlichen Radium-Aktivitäten auch dann unzertrennlich sind, wenn folgender „Kreisprozess“ durchgeführt wird.
Aus einem lang bestrahlten Uran wird der 12d-Körper RaIV nach Zugabe von 3 g BaCl2 abgetrennt. Von diesem nun radioaktiven Bariumchlorid werden etwa 0,5 g zurückbehalten, die übrige Menge wird nacheinander in folgende chemische Verbindungen umgewandelt:
-
- Bariumsuccinat (bernsteinsaures Barium)
- Bariumnitrat
- Bariumcarbonat
- Bariumchlorid
- Barium-Ferri-Mannit (organische Barium-Eisen-Verbindung)
- Bariumchlorid
wobei die jeweilige Verbindung gefällt oder auskristallisiert wird und nach dem Abtrennen von der wässrigen Lösung in die nächstfolgende Verbindung überführt wird.
Sowohl vom zurückbehaltenen wie auch dem mehrfach umgewandelten Bariumchlorid werden dann jeweils gleiche Mengen abwechselnd unter demselben Zählrohr gemessen. Über einen Zeitraum von 14 Tagen haben beide Präparate zum gleichen Zeitpunkt immer die gleiche Aktivität. An dem Mischungsverhältnis zwischen Barium und RaIV hat sich während des Kreislaufs also nichts geändert, RaIV verhält sich auch bei diesem Versuch wie Barium.
Der chemische Nachweis weiterer Spaltprodukte
Radioaktive Edelgase
Wenn der Urankern mit seinen 92 Kernladungen einen Bariumkern mit der Kernladung 56 abspaltet, bleibt ein Element mit der Kernladung 36 übrig, also ein Isotop des Edelgases Krypton. Da eine direkte Messung radioaktiver Edelgase schwierig erscheint, benutzen Hahn und Straßmann indirekte Nachweise anhand der Zerfallsprodukte des Kryptons, nämlich der radioaktiven Elemente 37 und 38 (Rubidium und Strontium). Am einfachsten lässt sich Strontium isolieren. Es gehört ebenso wie Barium zur Gruppe der Erdalkalimetalle, lässt sich jedoch leicht von Barium trennen, da Strontiumchromat im Gegensatz zu Bariumchromat in verdünnten Säuren löslich ist. Ein entsprechender Versuch bestätigt die Vermutung: Es wird radioaktives Strontium gefunden und auch dessen Folgeprodukt, ebenfalls ein β−-Strahler.
In einem zweiten Experiment beweisen sie auf einem ganz anderen Weg, dass ein Edelgas vorliegt und in ein Alkalielement zerfällt. Dazu bestrahlen sie eine wässrige Lösung von Uranylnitrat und lassen während der Bestrahlung einen Luftstrom hindurchperlen, um gebildete Edelgase (Krypton oder Xenon) auszutreiben. Der Luftstrom passiert anschließend ein zweites Gefäß mit Wasser, das die aus dem Edelgas entstehenden wasserlöslichen Zerfallsprodukte (Rubidium oder Cäsium) aufnimmt. Aus dieser Lösung werden dann die schweren Alkalielemente gemeinsam mit inaktivem Cäsiumträger ausgefällt.
In der Veröffentlichung [10] bleibt noch offen, ob es sich um Krypton-Rubidium oder Xenon-Cäsium handelt. Wenig später stellt sich heraus, dass beide Elementpaare vorliegen.
Der chemische Nachweis einer Kernspaltung des Thoriums
Der Bericht [10] bringt nicht nur den „endgültigen Beweis für das Entstehen von Barium aus dem Uran“, sondern auch den Nachweis, dass bei der Bestrahlung des Thoriums mit Neutronen ebenfalls Bariumisotope entstehen.
In ihrer vorhergehenden Veröffentlichung [9] hatten Hahn und Straßmann Bestrahlungsversuche an Thorium erwähnt, die sie 1938 noch gemeinsam mit Lise Meitner durchgeführt hatten und bei denen damals auch „Radium“-Isotope gefunden worden waren[8], die aus jetziger Sicht ebenfalls radioaktive Bariumisotope sein könnten. Die nun erfolgte, erneute Überprüfung (mit Thorium X als Indikator für Radium und einer Fraktionierung in Form der Chromate) bestätigt diese Vermutung.
Die physikalische Deutung der Kernspaltung
Bereits vor dem Abschluss des ersten Manuskripts zu[9], am 19. Dezember 1938, hatte Otto Hahn Lise Meitner brieflich über diese Ergebnisse informiert und sie gefragt, ob sie für das beobachtete Verhalten des Urans („Zerplatzen in Barium?“) irgendeine kernphysikalische Erklärung hätte. Ohne ihre Antwort abzuwarten, schreibt er ihr zwei Tage später: „Wir können unsere Ergebnisse nicht totschweigen, auch wenn sie physikalisch vielleicht absurd sind.“ Sie antwortet ihm auf seinen ersten Brief am 21. Dezember 1938:
„Mir scheint die Annahme eines so weitgehenden Zerplatzens sehr schwierig, aber wir haben in der Kernphysik so viele Überraschungen erlebt, dass man auf nichts ohne weiteres sagen kann: es ist unmöglich.“
Theoretische Überlegungen
Lise Meitner verbringt die Weihnachtstage bei schwedischen Freunden in der kleinen Stadt Kungälv in der Nähe von Göteborg und hat ihren Neffen Otto Frisch gebeten, sie dort zu besuchen. Frisch ist Physiker bei Niels Bohr am Institut für Theoretische Physik in Kopenhagen. Es wird nach seinen eigenen Worten der bedeutungsvollste Besuch seines Lebens, denn innerhalb weniger Tage finden sie gemeinsam die Lösung des Problems.
Von einem Atomkern waren bisher niemals größere Bruchstücke als Protonen oder Heliumkerne (Alphateilchen) abgespalten worden, eine Emission noch größerer Teilchen war nach wohl fundierten theoretischen Konzepten der Kernphysik äußerst unwahrscheinlich. Dieser Ansicht widersprechen jedoch die jüngsten Analysenergebnisse aus Berlin.
Meitner und Frisch greifen deshalb auf eine Modellvorstellung zurück, bei der Atomkerne mit elektrisch geladenen Wassertröpfchen verglichen werden. In diesem Konzept ist der große Urankern ein schon ziemlich labiles Gebilde, das – so ihr Schluss – durch das Einfangen eines Neutrons so heftig in Schwingungen geraten kann, dass er sich in zwei kleinere, etwa gleichgroße Kerne teilt. Diese beiden Atomkerne stoßen sich wegen ihrer positiven Kernladungen stark voneinander ab. Die dadurch frei werdende Energie schätzen sie auf etwa 200 Millionen Elektronenvolt (= 200 MeV); das ist weit mehr, als ein anderer Prozess irgendwelcher Art liefern könnte.
Meitner und Frisch bezeichnen diesen bisher unbekannten Vorgang als „fission process“ (in Anlehnung an den biologischen Vorgang der Zellteilung), im Deutschen werden daraus die Begriffe „Spaltprozess“ und „Kernspaltung“. Anschaulich beschrieben hat Frisch diese Überlegungen und Schlussfolgerungen, zu denen seine Tante und er bei einem Spaziergang im Schnee gelangt sind, in seinem Buch „Woran ich mich erinnere“ (vgl. Abschnitt „Sonstige Literatur“).
Über ihre Erkenntnisse berichten Meitner und Frisch in einer kurzen Notiz, die am 16. Januar 1939 bei der Zeitschrift „Nature“ eingereicht wird und am 11. Februar 1939 erscheint[11]. Otto Hahn erhält eine Kopie des Manuskripts am 24. Januar 1939.
Frisch fährt Anfang Januar nach Kopenhagen zurück. Als er Niels Bohr über die neuesten Ergebnisse aus Berlin und Kungälv informiert, schlägt dieser sich mit der Hand an die Stirn und ruft: „Ach, was für Idioten wir doch alle waren! Das hätten wir voraussehen können.“
Physikalische Bestätigung der Kernspaltung
Den theoretischen Überlegungen von Meitner und Frisch folgt eine Veröffentlichung von Otto Frisch, in der er das in [11] entworfene Konzept einer Spaltung in zwei energiereiche Fragmente experimentell bestätigt[12].
Frisch sucht nach den postulierten Fragmenten hoher Geschwindigkeit mit einer Kernladungszahl von etwa 40 bis 50 und einem Atomgewicht von etwa 100 bis 150. Trotz ihrer sehr hohen Energie von bis zu 100 MeV sollten diese Atomkerne nur eine Reichweite von einigen Millimetern haben, da sie aufgrund ihrer hohen Ladung die umgebende Luft sehr stark ionisieren und hierdurch rasch abgebremst werden. Solche Vorgänge lassen sich mit einer Ionisationskammer messen.
Frisch verwendet eine sehr kleine Ionisationskammer von nur 3 cm Durchmesser, in die er auf einem Kupferblech ein Uranpräparat einschiebt. Da die Reichweite der Fragmente von der Gasfüllung der Kammer abhängt (sie ist um so größer, je niedriger das Atomgewicht des Füllgases ist), füllt er sie mit Wasserstoff.
Die Ra-Be-Neutronenquelle (mit 300 mg Radium) ist so unter der Ionisationskammer angebracht, dass (zur Abbremsung der Neutronen) ein hohler Paraffinzylinder über die Neutronenquelle geschoben werden kann. Durch eine geeignete elektrische Schaltung mit einem Thyratron sorgt Frisch dafür, dass von dem angeschlossenen Zählwerk nur stark ionisierende Teilchen (mit einer Energie größer als 20 MeV) registriert werden; alle schwächeren Stromimpulse, vor allem die Alphateilchen des Urans (4,2 MeV), werden hingegen unterdrückt.
Bei der Bestrahlung mit ungebremsten Neutronen registriert die Messapparatur etwa 15 Spaltprodukte pro Minute; werden die Neutronen durch die Paraffinschicht abgebremst, so werden doppelt so viele dieser energiereichen Bruchstücke gemessen. Bei der Bestrahlung von Thorium sind die Ergebnisse ähnlich, langsame Neutronen verstärken hier den Effekt jedoch nicht. Mit diesem Versuch (durchgeführt am 13. und 14. Januar 1939) gelingt es Frisch, die Spaltung von Uran und Thorium auf physikalischem Wege nachzuweisen und die postulierte, ungewöhnlich hohe Energiefreisetzung zu bestätigen.
Ergänzende Beobachtungen zur Kernspaltung
Durch das Erscheinen der Berichte [9] [10] [11] [12] im Januar bzw. Februar 1939 wird weltweit eine wahre Flut von Untersuchungen ausgelöst. Bereits Ende Februar sind mehr als 40 Arbeiten erschienen, in denen die Kernspaltung des Urans durch Neutronen ebenfalls nachgewiesen wird.
Die Kettenreaktion beim Uranisotop 235
Niels Bohr entwickelt das von Meitner und Frisch vorgeschlagene Konzept weiter und kommt zu der Erkenntnis, dass die Spaltung des Urans nicht am Hauptisotop 238U, sondern am seltenen Uranisotop 235U erfolgt, das im natürlichen Uran nur zu 0,7 % vorhanden ist[13]. Eine Arbeitsgruppe um Frédéric Joliot findet, dass bei der Uranspaltung zwei bis drei Neutronen pro Spaltung freigesetzt werden[14] [15]. Damit ist die Möglichkeit für eine Kettenreaktion gegeben, in der diese Neutronen weitere Urankerne spalten.
Siegfried Flügge prüft in einer ausführlichen Abhandlung, ob und wie der Energieinhalt der Atomkerne in einer solchen „Uranmaschine“ technisch nutzbar gemacht werden kann[16].
Spaltprodukt-Paare
In ihrem Bericht [10] haben Hahn und Straßmann auch einige Vermutungen über die Massenzahl der Bariumisotope geäußert; danach dürfte BaIII das Atomgewicht 139 und BaIV das Atomgewicht 140 haben. Beide sind Isotope, die im natürlich vorkommenden Barium nicht vorhanden sind.
Unterstellt man nun, dass bei der Uranspaltung zusammen mit jedem der beiden ebengenannten Bariumisotope sowohl zwei als auch drei freie Neutronen auftreten können, so ergeben sich allein für diese beiden Bruchstücke folgende Spaltprozesse:
Dass die Spaltung jedoch nicht auf die Kombination Barium + Krypton beschränkt ist, haben Hahn und Straßmann bereits in ihrem Bericht angedeutet, einige Wochen später können sie dann auch die Elementkombination Xe + Sr als weiteres Spaltproduktpaar nachweisen[17], schließlich auch Cs + Rb[18].
Betrachtet man die ebengenannten und noch weitere Spaltpaare in der geordneten Reihenfolge
so erkennt man, dass sowohl die erstgenannten Partner (I……La) als auch die zweiten (Y……Br) jeweils aufeinanderfolgende Elemente im periodischen System sind, wobei sich beide Gruppen in ihrer Massenzahl deutlich voneinander unterscheiden. Die Uranspaltung erfolgt also asymmetrisch; wie sich in den folgenden Jahren herausstellt, gruppieren sich die schweren Partner um die Massenzahl 138, die leichten um die Masse 95. Nimmt man die selteneren Produkte hinzu, so sind im Spaltproduktgemisch um die vierzig Elemente, vom Kupfer bis zu den schweren Lanthaniden vertreten und von jedem dieser Elemente mehrere Isotope.
Spaltprodukt-Zerfallsketten
Die primären Spaltprodukte, die unmittelbar nach dem Neutroneneinfang eines Urankerns durch dessen Spaltung entstehen, wandeln sich durch β−-Zerfall um und bilden Zerfallsketten, bis ein stabiler Kern erreicht ist. Aus einem primären Spaltprodukt Lanthan z. B. entsteht die Zerfallskette
-
- La → Ce → Pr → Nd (stabil)
aus einem primären Barium eine andere Kette
-
- Ba → La → Ce → Pr (stabil)
Reaktionsprodukte der Bestrahlung entstehen also sowohl primär durch Spaltung als auch sekundär durch Zerfall.
Bei den Versuchen in Rom, Paris und Berlin waren alle diese Spaltprodukte im neutronenbestrahlten Uran vorhanden, die Beteiligten haben jedoch nicht gezielt danach gesucht!
Der 3,5h-Körper von Curie und Savitch wird im Nachhinein als ein Isotopengemisch identifiziert, das hauptsächlich aus dem Spaltprodukt Yttrium-92 (HWZ: 3,5 h) besteht, einem Isotop des chemischen Elements Yttrium, das in der gleichen Gruppe des periodischen Systems steht wie Lanthan.
Die Äquivalenz von Masse und Energie
Bereits 1905 folgerte Albert Einstein aus seiner speziellen Relativitätstheorie, dass Masse und Energie ineinander übergeführt werden können.
-
- E = δm • c2 (c = Lichtgeschwindigkeit)
Diese Gleichung gilt für jeden chemischen oder physikalischen Vorgang, bei dem Energie E (zumeist in Form von Wärme) auftritt oder verbraucht wird; die hierbei auftretenden Massenänderungen δm sind jedoch bei normalen Energieprozessen (Verbrennung) so gering, dass sie experimentell nicht gemessen werden können.
Energie und Massenänderung bei der Kernspaltung
Atommassen der an einer Uranspaltung beteiligten Reaktionspartner gemäß „Table of the Nuclides“, Rutherford Laboratory: Isotop Masse
des Isotops
(amu)Neutron 1,008664 235U 235,043922 139Ba 138,908835 140Ba 139,910599 93Kr 92,931265 94Kr 93,934362 95Kr 94,939840 Die Ursache für die ungeheure Energiefreisetzung bei der Kernspaltung ist eine Umwandlung von Masse in Energie, die auf Grund der Eigenschaften der Atomkerne eintritt: Die Masse eines Atomkerns ist immer geringer als die Summe der Massen seiner Bausteine (Protonen und Neutronen = Nukleonen). Dieser Massendefekt ist die „Bindungsenergie“ des Atomkerns; sie würde freigesetzt, wenn man ihn aus seinen Protonen und Neutronen zusammensetzen würde. Die Bindungsenergie wächst mit der Zahl der Nukleonen im Kern, jedoch nicht gleichmäßig, sondern sie ist pro Nukleon gerechnet für mittelschwere Atomkerne wie z. B. die Spaltprodukte deutlich größer als für die Atomkerne des Urans. Dieser Überschuss an Bindungsenergie wird beim Spaltprozess frei, wenn aus einem Uran- zwei Spaltproduktkerne entstehen.
Aus den heute bekannten exakten Werten für die Atommassen der Reaktionspartner lässt sich die bei der Kernspaltung auftretende Massenänderung einfach ermitteln und hieraus dann die freiwerdende Energie berechnen.
Aus diesen Werten ergibt sich für die Kernreaktion
-
- 235U + n → 139Ba + 94Kr + 3 n
eine Massenänderung von 0,183 amu; um so viele Masseneinheiten sind die Spaltprodukte auf der rechten Seite der Gleichung leichter als das Uranisotop 235 samt Neutron. Bei der ähnlichen Kernreaktion
-
- 235U + n → 140Ba + 94Kr + 2 n
ist der Massendefekt mit 0,190 amu sogar noch etwas größer. Von der ursprünglich vorhandenen Masse von etwa 236 amu sind also 0,183 amu bzw. 0,19 amu verschwunden; dieser Massenanteil von etwa 0,08 Prozent ist als Energie freigesetzt worden, deren Größe nach der von Einstein angegebenen Gleichung errechnet werden kann. Da der Masse von 1 amu eine Energie von 931,494 MeV entspricht, ergeben 0,19 amu Massendefekt etwa 177 MeV Energiegewinn.
Meitner und Frisch standen die exakten Daten für die Atommassen nicht zur Verfügung, sie haben deshalb die kinetische Energie der beiden Bruchstücke des Urankerns (die sich wegen ihrer positiven Kernladungen gegenseitig abstoßen) aus dem Coulombschen Gesetz errechnet[11], wobei sie für den Abstand der beiden kugelförmigen Spaltprodukte die Summe der Kernradien einsetzen mussten, deren Größe sie nur abschätzen konnten. Sie lagen mit den von ihnen errechneten 200 MeV jedoch bemerkenswert gut.
Kernenergie – Konventionelle Energie; ein Vergleich
Energieumrechnungstabellen kann man weiterhin entnehmen, dass einem Massenverlust von 1 amu eine Energie von 1,492 • 10−10 Joule entspricht, für 0,19 amu sind dies also etwa 0,3 • 10−10 Joule. Verglichen mit der Wärmemenge, die bei einer normalen chemischen Reaktion auftritt, z. B.
-
- C + O2 —> CO2 + 393 000 Joule
scheint die bei der Kernspaltung freiwerdende Energie demnach äußerst gering zu sein!
Der Grund für diese vermeintliche Diskrepanz ist das in Kernphysik und Chemie unterschiedliche Bezugssystem bei der Angabe und Berechnung von Energieänderungen während einer chemischen oder kernphysikalischen Reaktion. In der Chemie sind Reaktionsgleichungen immer Mol-Gleichungen, jedes chemische Symbol in einer solchen Gleichung steht für ein Mol des betreffenden chemischen Elements oder Moleküls, die Energieangaben (in Joule) gelten für diese Molmengen. Bei Kernreaktionen wird die Energie in MeV angegeben; sie bezieht sich immer auf den atomaren Einzelprozess, hier also auf die Spaltung eines einzigen Atomkerns.
Will man also beide Angaben miteinander vergleichen, so muss man zuvor den Massenverlust bei der Kernspaltung von einem Mol Uran-235 berechnen. Mithilfe der Loschmidtschen Zahl L ergibt sich hierfür ein Wert von L • 0,19 amu = 1,14 • 1023 amu; dies entspricht dann einer Energie von 1,7 • 1013 Joule.
Unterstellt man, dass die bei beiden Reaktionen freiwerdende Energie mit einem Wirkungsgrad von 100 % in Wärme umgesetzt und zum Erwärmen von Wasser bis zu dessen Siedepunkt genutzt werden könnte, so kann man durch die Verbrennung von 12 Gramm Kohlenstoff etwa 1 Liter Wasser zum Kochen bringen, bei der Kernspaltung von 235 Gramm Uran-235 sind es 5 • 107 Liter Wasser (= 50.000 Kubikmeter)!
Die tatsächlichen Transurane
Mit der Entdeckung der Kernspaltung war die Interpretation aller zuvor in Berlin durchgeführten Versuche fragwürdig geworden. Die vermeintlichen Transurane erwiesen sich nun als unterschiedliche Spaltprodukte des Urans. Damit begann die Suche nach den Transuranen (siehe auch Isotopentabelle) von neuem.
Element 93: Neptunium
Ausgangspunkt ist der 23-Minuten-Körper, den das KWI-Team 1937 gefunden und einwandfrei als Uranisotop 239 identifiziert hatte (vgl. Abschnitt Ein Transuran unter den Reaktionsprodukten).
Als β−-Strahler muss dieses Isotop in Element 93 zerfallen; alles Suchen danach in den Mitfällungen der schwer löslichen Sulfide blieb indessen in Berlin erfolglos.
Die Entdeckung von Element 93 und die Identifizierung gerade dieses Isotops 239 von Element 93 gelingt 1940 in Berkeley (USA) durch E. McMillan und P. H. Abelson. Als Neutronenquelle verwenden sie das dort vorhandene, damals weltweit größte Cyclotron. Mithilfe einer Be-D-Reaktion (Beschuss eines Berylliumpräparats mit Deuteronen)
erzeugt es einen Neutronenstrom, der dem einer Radium-Beryllium-Neutronenquelle mit etwa 1200 kg Radium entspricht.
Nach dem intensiven Beschuss einer dünnen Uranschicht kann an dem Präparat ohne eine vorhergehende chemische Trennung neben der 23min-Aktivität des 239U eine weitere β−-Strahlung mit einer HWZ von 2,3 Tagen gemessen werden, die von dem gesuchten Element 93 herrühren könnte:
Dieser Zusammenhang wird im Laufe der Untersuchungen chemisch eindeutig bewiesen. Anders als erwartet erweist sich Element 93 jedoch nicht als ein homologes Element zu Rhenium. Es wird nicht mit Platinsulfid zusammen ausgefällt, sondern verhält sich ganz anders, nämlich sehr ähnlich wie Uran. Damit ist auch klar, weshalb die Suche nach Element 93 in Berlin und anderswo scheiterte: an falschen Annahmen über die chemischen Eigenschaften dieses Elements.
McMillan und Abelson veröffentlichen ihre Entdeckung Mitte Juni 1940,[19] in Berlin wird sie Ende August bekannt. Der Name Neptunium wird später in Anlehnung an die sonnenfernen Planeten Uranus, Neptun und Pluto gewählt.
Hahn und Straßmann versuchen nun erneut, das Folgeprodukt des 23-Minuten-Körpers zu finden. Da die β−-Strahlung von 239Np jedoch sehr energiearm ist (um 0,4 MeV), gelingt ihnen dies erst nach dem Bau eines besonders dünnwandigen Zählrohres. Sie entwickeln ein chemisches Verfahren, um dieses Neptunium-Isotop aus großen Mengen von neutronenbestrahltem Uran zu isolieren. Die von McMillan und Abelson beschriebenen chemischen Eigenschaften können sie bestätigen und wesentlich ergänzen.[20] Auch sie kommen zu dem Ergebnis, dass das Element 93 kein homologes Element zu Rhenium sein kann, das Periodensystem der Elemente in der Anordnung von 1938 also korrekturbedürftig ist.
Element 94: Plutonium
Da Neptunium-239 ein β−-Strahler ist, muss es in ein Isotop 239 des Elements 94, das spätere Plutonium, zerfallen. McMillan und Abelson suchen deshalb in sehr starken Präparaten von Neptunium-239 nach diesem neuen Element, jedoch ohne Erfolg und mit dem Schluss, dass es sich um ein sehr langlebiges und somit selten strahlendes Isotop handeln müsse.[19]
Mit fortschreitender Kenntnis des Spaltprozesses wird klar, dass dieses Plutoniumisotop ähnlich gut spaltbar sein sollte wie Uran-235. Die Suche am Berkeley-Cyclotron wird deshalb, nun als geheime Forschung, intensiviert.
Man wird auf einem Umweg fündig. Bei der Bestrahlung von Uran mit Deuteronen wird von G. T. Seaborg, E. McMillan, J. W. Kennedy und A. C. Wahl folgende Kernreaktion beobachtet:[21]
Das Np-Isotop 238 zerfällt gemäß
in 238Pu, einen langlebigen α-Strahler, der seinerseits ein Uran-Isotop nachbildet. An 238Pu werden die chemischen Eigenschaften von Plutonium studiert. Es erweist sich als dem Neptunium und Uran eng verwandt und ist sehr schwierig von diesen Elementen zu trennen. Mit diesen Kenntnissen gelingt es aber dann, das gesuchte Plutoniumisotop 239 zu finden, einen Alphastrahler mit 24000 Jahren Halbwertszeit:[22]
239Pu ist, wie erwartet, sehr gut spaltbar durch Neutronen; auch dabei werden Neutronen emittiert, so dass auch bei diesem Element eine Kettenreaktion möglich ist.
McMillan und Seaborg werden für ihre Entdeckungen 1951 gemeinsam mit dem Nobelpreis für Chemie ausgezeichnet.
In Deutschland wird nicht nach dem Folgeprodukt von 239Np, also 239Pu, gesucht. Hahn und Straßmann, die sich intensiv mit dem Element 93 beschäftigten, mussten dies nach den vergeblichen Versuchen von McMillan und Abelson angesichts ihrer eigenen schwachen Neutronenquellen als aussichtslos ansehen.
Weitere Transurane
Die Elemente 95 (Americium) und 96 (Curium), die 1944/45 ebenfalls in den USA entdeckt wurden, setzen die uranähnlichen Eigenschaften nicht fort, sondern lehnen sich stark an das Actinium an. Daraus folgert Seaborg, dass im Periodensystem beim Actinium eine zu den Lanthanoiden homologe Reihe, die der Actinoiden beginnt, die sich bis zum Element 103 (Lawrencium) erstrecken sollte, eine Voraussage, die sich in den folgenden Jahrzehnten bestätigt hat.
Mit dem Element 104 beginnt dann die Reihe der Transactinoide. Gegenwärtig (2006) sind deren chemische Eigenschaften bis zum Element 108 einschließlich erforscht.[23] Im periodischen System steht jetzt unter dem Rhenium das Element 107 (Bohrium), mit chemischen Eigenschaften, die man in den 1930er Jahren für das Element 93 vermutet hatte.
Fazit
Rückblicke der an der Entdeckung der Kernspaltung unmittelbar beteiligten Personen[24][25][26] bringen übereinstimmend zum Ausdruck:
„Die Entdeckung der Kernspaltung ergab sich, ohne dass man nach ihr gesucht hätte, aus der jahrelangen konsequenten Verfolgung bisweilen unerwarteter radiochemischer Versuchsergebnisse.“
In der Kernphysik hatte man bei der Herstellung künstlich radioaktiver Isotope ebenso wie bei dem Zerfall natürlich radioaktiver Elemente bis zum Jahresende 1938 immer nur Kernumwandlungen beobachtet, bei denen die neu entstandenen Elemente im periodischen System ganz nahe zum Ausgangselement standen. Bei dem Wettlauf um die Synthese der ersten Transurane aus Uran ging man in Rom, Berlin und Paris deshalb davon aus, die beobachteten radioaktiven Substanzen könnten nichts anderes als Isotope des Urans oder seiner nächsten Nachbarn sein, rechts oder allenfalls auch links von ihm im periodischen System. Zudem nahm man an, dass die Transurane chemisch dem Rhenium und den Platinmetallen ähnlich sind.
Eine bereits 1934 von Ida Noddack geäußerte Vermutung[27]: „Es wäre denkbar, daß bei der Beschießung schwerer Kerne mit Neutronen diese Kerne in mehrere größere Bruchstücke zerfallen, die zwar Isotope bekannter Elemente, aber nicht Nachbarn der bestrahlten Elemente sind“ blieb deshalb sowohl in Rom wie auch in Berlin und Paris unbeachtet; auf die weitere Entwicklung hatte sie keinen Einfluss.
Irène Curie und Paul Savitch kamen im Sommer 1938 der Lösung allerdings schon näher, indem sie postulierten, bei den Neutronenbestrahlungen von Uran ein Transuran mit ganz anderen chemischen Eigenschaften gefunden zu haben. Dies veranlasst Otto Hahn und Fritz Straßmann, nach Radiumisotopen zu suchen und sie finden gleich mehrere. Um diesen Befund zu erhärten, überprüfen sie deren chemisches Verhalten im Detail, mit einem völlig unerwarteten Ergebnis: Die vermeintlichen Radiumisotope sind radioaktive Bariumisotope; der Urankern spaltet sich nach Einfang eines Neutrons in leichtere Elemente.
Dieses Ergebnis veröffentlichen Hahn und Straßmann Anfang 1939 „nur zögernd“, doch können sie innerhalb weniger Wochen weitere chemische Beweise vorlegen. Gleichzeitig finden die hinsichtlich einer plausiblen Erklärung um Hilfe gebetenen Kernphysiker Lise Meitner und Otto Frisch ein qualitatives Modell für den neuartigen Prozess und Frisch gelingt die erste Bestätigung durch ein physikalisches Experiment.
Die Resonanz ist außerordentlich. Bis Dezember 1939 erscheinen bereits mehr als 100 Publikationen, die sich mit dem Problem der Kernspaltung befassen[28].
Weit über die Wissenschaften hinaus reichende Konsequenzen erhält die Kernspaltung jedoch dadurch, dass bei jedem Spaltprozess neben den beiden Bruchstücken auch noch einige Neutronen freigesetzt werden, die in anderen Urankernen weitere Spaltvorgänge auslösen. Diese Kettenreaktion ermöglicht es, die extrem hohe Spaltungsenergie technisch zu nutzen. Bis zur Verwirklichung dieser Idee dauert es nur drei Jahre. Der erste funktionsfähige Atomreaktor, von Enrico Fermi an der Universität in Chicago erbaut, wird im Dezember 1942 „kritisch“, das heißt, die Kettenreaktion läuft selbständig, der Reaktor produziert Energie. Im Juli 1945 wird dann aber auch die erste Atombombe gezündet (in der Wüste von Alamogordo/New Mexico); der Januskopf der Kernspaltung tritt damit zutage.
Ehrungen
1945 Verleihung des Nobelpreises für Chemie für das Jahr 1944 an Otto Hahn (Überreichung im Dezember 1946) 1966 Verleihung des Enrico-Fermi-Preises an Lise Meitner, Otto Hahn und Fritz Straßmann Einzelnachweise
- ↑ E. Fermi: Possible Production of Elements of Atomic Number Higher than 92. Nature 133 (1934) 898–899
- ↑ S. Curie: Untersuchungen über die radioaktiven Substanzen. Dissertation von Marie Curie; übersetzt von W. Kaufmann. Die Wissenschaft, Erstes Heft, S. 23–33. Braunschweig: Vieweg (1904)
- ↑ L. Meitner, O. Hahn, F. Straßmann: Über die Umwandlungsreihen des Urans, die durch Neutronenbestrahlung erzeugt werden. Zeitschrift für Physik 106 (1937) 249–270
- ↑ O. Hahn, L. Meitner, F. Straßmann: Über die Transurane und ihr chemisches Verhalten. Berichte der Deutschen Chemischen Gesellschaft 70 (1937) 1374–1392
- ↑ O. Hahn, L. Meitner, F. Straßmann: Ein neues langlebiges Umwandlungsprodukt in den Trans-Uranreihen. Die Naturwissenschaften 26 (1938) 475–476
- ↑ a b I. Curie, P. Savitch: Sur les radioéléments formés dans l'uranium irradié par les neutrons II. Le Journal de Physique et le Radium 9 (1938) 355–359
- ↑ O. Hahn, F. Straßmann: Über die Entstehung von Radiumisotopen aus Uran durch Bestrahlen mit schnellen und verlangsamten Neutronen. Die Naturwissenschaften 26 (1938) 755–756
- ↑ a b L. Meitner, F. Straßmann, O. Hahn: Künstliche Umwandlungsprozesse bei Bestrahlung des Thoriums mit Neutronen; Auftreten isomerer Reihen durch Abspaltung von α-Strahlen. Zeitschrift für Physik 109 (1938) 538–552
- ↑ a b c d O. Hahn, F. Straßmann: Über den Nachweis und das Verhalten der bei der Bestrahlung des Urans mittels Neutronen entstehenden Erdalkalimetalle. Die Naturwissenschaften 27 (1939) 11–15
- ↑ a b c d e O. Hahn, F. Straßmann: Nachweis der Entstehung aktiver Bariumisotope aus Uran und Thorium durch Neutronenbestrahlung; Nachweis weiterer aktiver Bruchstücke bei der Uranspaltung. Die Naturwissenschaften 27 (1939) 89–95
- ↑ a b c d L. Meitner, O. R. Frisch: Disintegration of Uranium by Neutrons: A New Type of Nuclear Reaction. Nature 143 (1939) 239–240.
- ↑ a b O. R. Frisch: Physical Evidence for the Division of Heavy Nuclei under Neutron Bombardment. Nature 143 (1939) 276–277
- ↑ N. Bohr: Resonance in Uranium and Thorium Disintegrations and the Phenomenon of Nuclear Fission. Physical. Review 55 (1939) 418–419
- ↑ H. von Halban, F. Joliot, L. Kowarski: Liberation of Neutrons in the Nuclear Explosion of Uranium. Nature 143 (1939), 470–471
- ↑ H. von Halban, F. Joliot, L. Kowarski: Number of Neutrons Liberated in the Nuclear Fission of Uranium. Nature 143 (1939), 680
- ↑ S. Flügge: Kann der Energieinhalt der Atomkerne technisch nutzbar gemacht werden? Die Naturwissenschaften 27 (1939) 402–410
- ↑ O. Hahn, F. Straßmann: Über die Bruchstücke beim Zerplatzen des Urans. Die Naturwissenschaften 27 (1939) 163
- ↑ O. Hahn, F. Straßmann: Weitere Spaltprodukte aus der Bestrahlung des Urans mit Neutronen. Die Naturwissenschaften 27 (1939) 529–534
- ↑ a b E. McMillan und P. H. Abelson: Radioactive Element 93. Physical Review 57 (1940) 1185–1186; doi:10.1103/PhysRev.57.1185.2
- ↑ F. Straßmann, O. Hahn: Über die Isolierung und einige Eigenschaften des Elements 93. Die Naturwissenschaften 30 (1942) 256–260
- ↑ G. T. Seaborg, E. McMillan, J. W. Kennedy, A. C. Wahl: Radioactive Element 94 from Deuterons on Uranium. Physical Review 69 (1946) 366–367; doi:10.1103/PhysRev.69.367.
- ↑ J. W. Kennedy, G. T. Seaborg, E. Segrè, A. C. Wahl: Properties of Element 94. Physical Review 70 (1946) 555–556; doi:10.1103/PhysRev.70.555.
- ↑ M. Schädel: Chemie superschwerer Elemente. Angewandte Chemie 118 (2006) 378–414
- ↑ O. Hahn: Die Auffindung der Uranspaltung. FIAT Review of German Science 1939–1946, Nuclear Physics and Cosmic Rays, Part 1. Dieterich, Wiesbaden (1948) S. 171–178
- ↑ L. Meitner: Wege und Irrwege zur Kernenergie. Naturwissenschaftliche Rundschau 16 (1963) 167–169
- ↑ F. Straßmann: Kernspaltung – Berlin, Dezember 1938 Privatdruck, Mainz 1978
- ↑ I. Noddack: Über das Element 93. Angewandte Chemie 47 (1934) 653–655
- ↑ L. A. Turner: Nuclear Fission. Reviews of Modern Physics 12 (1940) 1–29
Sonstige Literatur
Bücher
- Hahn, Otto: Vom Radiothor zur Uranspaltung. Braunschweig: Friedr. Vieweg & Sohn, 1962. Nachdruck 1989
- Wohlfarth, Horst: 40 Jahre Kernspaltung – Eine Einführung in die Originalliteratur. 'Darmstadt: Wissenschaftliche Buchgesellschaft, 1979; ISBN 3-534-08206-0
- Frisch, Otto Robert: Woran ich mich erinnere – Physik und Physiker meiner Zeit. Stuttgart: Wissenschaftliche Verlagsgesellschaft mbH, 1981; ISBN 3-8047-0614-2
- Krafft, Fritz: Im Schatten der Sensation – Leben und Wirken von Fritz Straßmann. Weinheim: Verlag Chemie, 1981; ISBN 3-527-25818-3
- Gerlach, Walther und Hahn, Dietrich: Otto Hahn - Ein Forscherleben unserer Zeit. Stuttgart: Wissenschaftliche Verlagsgesellschaft mbH, 1984; ISBN 3-8047-0757-2
- Brommer, Peter und Herrmann, Günter: Fritz Straßmann (1902–1980) – Mitentdecker der Kernspaltung. Inventar des Nachlasses und Kommentierung der Versuche zur Kernspaltung. Veröffentlichungen der Landesarchivverwaltung Rheinland-Pfalz Band 95. Koblenz: Verlag der Landesarchivverwaltung Rheinland-Pfalz, 2001; ISBN 3-931014-57-6
- Meitner, Lise: Erinnerungen an Otto Hahn. Stuttgart: S. Hirzel Verlag, 2005; ISBN 3-7776-1380-0
Zeitschriften
- H. Menke, G. Herrmann: Was waren die Transurane der dreißiger Jahre in Wirklichkeit? Radiochimica Acta 16 (1971) 119–123
- W. Seelmann-Eggebert: Über die Entdeckung der Kernspaltung. Ein historischer Rückblick. Chimia 33 (1979) 275–282
- F. Krafft: Ein frühes Beispiel interdisziplinärer Team-Arbeit. Zur Entdeckung der Kernspaltung durch Hahn, Meitner und Straßmann. Physikalische Blätter 36 (1980) 85–89, 113–118.
- P. Brix: Die folgenreiche Entdeckung der Uranspaltung – und wie es dazu kam. Physikalische Blätter 45 (1989) 2–10
- G. Herrmann: Vor fünf Jahrzehnten: Von den „Transuranen“ zur Kernspaltung. Angewandte Chemie 102 (1990) 469–496
- G. Herrmann: The Discovery of Nuclear Fission – Good Solid Chemistry Got Things on the Right Track. Radiochimica Acta 70/71 (1995) 51–67
Weblinks
-

![{}^{109}_{\ 47} \mathrm {Ag} + {}^{1}_{0} \mathrm {n} \ \rightarrow \ {}^{110}_{\ 47} \mathrm {Ag} \ \xrightarrow[\text{24 s}]{\text{ }\mathrm{\beta}\mathrm{^-}} \ {}^{110}_{\ 48} \mathrm {Cd}](/pictures/dewiki/50/28b039efc4b926d5f9c0003b1d698dc0.png)
![{}^{238}_{\ 92} \mathrm {U} + {}^{1}_{0} \mathrm {n} \ \rightarrow \ {}^{239}_{\ 92} \mathrm {U} \ \xrightarrow[\text{ }]{\text{ }\mathrm{\beta}\mathrm{^-}} \ {}^{239}_{\ 93} \mathrm {Element}](/pictures/dewiki/102/f6b017b67775aa248d89620d33e70369.png)
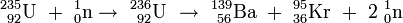
![{}^{238}_{\ 92} \mathrm {U} \ \xrightarrow[{4,5} \ \cdot \ {10^9} {\ a}] {\text{ }\mathrm{\alpha}} \ {}^{234}_{\ 90} \mathrm {Th \ (UX1)} \ \xrightarrow[\text{ 24,1 d }]{\text{ }\mathrm{\beta}\mathrm{^-}} \ {}^{234}_{\ 91} \mathrm {Pa \ (UX2)} \ \xrightarrow[\text{ 1,17 m }]{\text{ }\mathrm{\beta}\mathrm{^-}} \ {}^{234}_{\ 92} \mathrm {U} \ \xrightarrow[{2,5} \ \cdot \ {10^5} {\ a}] {\text{ }\mathrm{\alpha}} \ {}^{230}_{\ 90} \mathrm {Th}](/pictures/dewiki/97/abfbe4fd90736c7b38072adc7a0d8977.png)
![{}^{238}_{\ 92} \mathrm {U} + {}^{1}_{0} \mathrm {n} \ \rightarrow \ {}^{239}_{\ 92} \mathrm {U} \ \xrightarrow[\text{ 23 m }]{\text{ }\mathrm{\beta}\mathrm{^-}} \ {}^{239}_{\ 93} \mathrm {EL93}](/pictures/dewiki/102/f282c3b0e2e4f5fb6bd3ee57dd048d65.png)
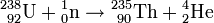
![{}^{231}_{\ 88} \mathrm {Ra}\ \xrightarrow[\text{ }]{\text{ }\mathrm{\beta}\mathrm{^-}}\ {}^{231}_{\ 89} \mathrm {Ac}](/pictures/dewiki/101/e7cca3d9016b4a207d10cbd6a73981b6.png)
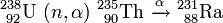
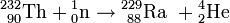
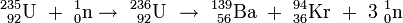
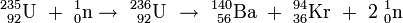
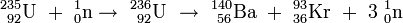
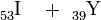
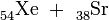
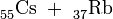
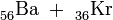
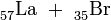
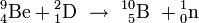
![{}^{238}_{\ 92} \mathrm {U} + {}^{1}_{0} \mathrm {n} \ \rightarrow \ {}^{239}_{\ 92} \mathrm {U} \ \xrightarrow[\text{ 23 min}]{\text{ }\mathrm{\beta}\mathrm{^-}} \ {}^{239}_{\ 93} \mathrm {EL93}\ \xrightarrow[\text{ 2,3 d}]{\text{ }\mathrm{\beta}\mathrm{^-}} \ {}^{239}_{\ 94} \mathrm {EL94}](/pictures/dewiki/101/e2cdf3ab065304465aef4e61029a8a51.png)
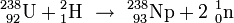
![{}^{238}_{\ 93} \mathrm {Np} \ \xrightarrow[\text{ 2,1 d }]{\text{ }\mathrm{\beta}\mathrm{^-}} \ {}^{238}_{\ 94} \mathrm {Pu} \ \xrightarrow[\text{ 88 a }]{\text{ }\mathrm{\alpha}} \ {}^{234}_{\ 92} \mathrm {U}](/pictures/dewiki/50/23a383f6055f8043618a072bd1aad026.png)
![{}^{238}_{\ 92} \mathrm {U} \ + \ {}^{1}_{0} \mathrm {n} \rightarrow \ {}^{239}_{\ 92} \mathrm {U} \ \xrightarrow[\text{23 min}]{\text{ }\mathrm{\beta}\mathrm{^-}} \ {}^{239}_{\ 93} \mathrm {Np} \ \xrightarrow[\text{2,33 d}]{\text{ }\mathrm{\beta}\mathrm{^-}} \ {}^{239}_{\ 94} \mathrm {Pu} \ \xrightarrow[\text{24000 a}]{\text{ }\mathrm{\alpha}} \ {}^{235}_{\ 92} \mathrm {U}](/pictures/dewiki/99/c88469ed42d020e303653c482809be78.png)
Wikimedia Foundation.