- Haplotype
-
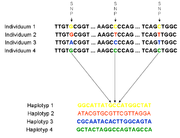 Haplotypen aus SNPs von Chromosomenabschnitten des gleichen Chromosoms von vier haploiden Individuen.
Haplotypen aus SNPs von Chromosomenabschnitten des gleichen Chromosoms von vier haploiden Individuen.Als Haplotyp (von griechisch haplús oder haplóos = einfach und typos = typ), eine Abkürzung von haploider Genotyp, wird eine Variante einer Nukleotidsequenz auf ein und demselben Chromosom im Genom eines Lebewesens bezeichnet. Ein bestimmter Haplotyp kann individuen-, populations- oder auch artspezifisch sein.
Die dabei verglichenen Allele können, wie beim International HapMap Project, individuelle Kombinationen von SNPs sein, die als genetische Marker benutzt werden können.[1]
Inhaltsverzeichnis
Geschichte
Der Begriff wurde 1967 von R. Ceppellini eingeführt.[2] Er wurde ursprünglich dazu benutzt, die genetische Zusammensetzung des MHC zu beschreiben, einem Komplex von Genen, der für das Immunsystem wichtige Proteine kodiert.
Abgrenzung zum Genotyp
Besitzt ein diploider Organismus bezüglich zweier Allele A und B den Genotyp AaBb, so können dem die Haplotypen AB|ab oder Ab|aB zugrunde liegen. Im ersteren Fall besitzt ein Chromosom die Allele A und B, das andere a und b. Im letzteren Fall besitzt ein Chromosom die Allele A und b, das andere a und B.
Bestimmung von Haplotypen
Zwei Fälle können unterschieden werden (im Folgenden bezieht sich der Begriff "Allel" auf die unterschiedlichen Nukleotide A,C,G und T):
- Die Bestimmung der Haplotypen einer Population haploider Individuen aus derselben Spezies (z.B. verschiedene E. coli Stämme) ist trivial. Hierfür ist die Sequenzierung und Bestimmung der SNPs der gegebenen Population ausreichend (siehe Bild). Werden Individuen bei der Sequenzierung ausgelassen so können andere darin enthaltene Allele (und die sich daraus ergebenden SNPs) natürlich nicht erfasst werden.
- 2. Polyploide Spezies
- Ist der Ploidiegrad der betrachteten Spezies grösser/gleich 2 so kompliziert sich das Problem (z.B. ist Homo sapiens diploid, Kartoffel tetraploid und Weichweizen hexaploid). In diesem Fall wird das Genom aus zwei oder mehr homologen Chromosomensätzen zusammengesetzt, wobei in der Regel die eine Hälfte vom mütterlichen und die andere vom väterlichen Elternteil stammt. Es müssen verschiedene Arten von SNPs unterschieden werden:
- 2.1 Wenn sich in einem Individuum ein mütterlicher und ein väterlicher homologer Chromosomen Satz in Nukleotidpositionen der DNA unterscheiden so werden diese SNPs bei Sequenzierung der entsprechenden Chromosomen des Individuums sichtbar (es wird immer eine Mischung der homologen Chromosomen sequenziert). Solch ein SNP wird im entsprechenden Individuum heterozygoter SNP genannt.[3]
- 2.2 Wenn in einem Individuum ein mütterlicher und ein väterlicher homologer Chromosomen Satz in einem betrachteten Locus identisch ist so werden bei Sequenzierung der DNA des Individuums keine SNPs sichtbar. Erst wenn bei mindestens einem zweiten Individuum im selben Locus ein anderes Allel gefunden wird kann an der entsprechenden Nukleotidposition von einem SNP gesprochen werden. Solch ein SNP wird im ersten Individuum homozygoter SNP genannt, kann aber in einem anderen Individuum einen heterozygoten SNP darstellen.[3][4]
- 2.3 Tauchen in einem SNP zwei verschiedene Allele auf (relativ zur gesamten betrachteten Population) so wird dieser SNP biallelisch genannt. Finden sich drei verschiedenen Allele so wird dieser SNP triallelisch und bei vier Allelen tetraallelisch genannt. Ein tetraallelischer SNP enthält die maximal Anzahl an verschiedenen Allelen da SNPs nur aus den vier Nukleotiden A,C,G und T gebildet werden können.[4]
- 2.4 Achtung: Diploide Spezies können prinzipiell tetraallelische SNPs besitzen obwohl für ein Individuum nur maximal zwei Allele möglich sind![4]
Wird nun in einer polyploiden Population (derselben Spezies) ein SNP bestimmt so lassen sich die Haplotypen (der Länge 1) wie in 1. direkt aus der Sequenzierung ablesen. Schon bei zwei SNPs wird es problematisch: bei der Sequenzierung geht die Zuordnung der einzelnen Allele zu ihren ursprünglichen Chromosomen verloren. Verschiedene Kombinationen der Allele in SNP 1 und SNP 2 sind nun möglich und damit auch verschiedene Haplotypen. Die Anzahl der möglichen Haplotypen wächst exponentiell mit der Anzahl der SNPs.
Verschiedene Methoden wurden entwickelt um Haplotypen in polyploiden Spezies zu bestimmen.
- i) Experimentell:
- Ein gegebenes Chromosom eines gegebenen Individuums wird mehrmals sequenziert und der entsprechende Haplotyp bestimmt. Bei jeder Sequenzierung wurde zufällig eines der homologen Chromosomen aus dem polyploiden Satz ausgewählt. Die Anzahl der Sequenzierungen wird so gewählt dass mit einer bestimmten Wahrscheinlichkeit davon ausgegangen werden kann dass kein Haplotyp bei der Sequenzierung ausgelassen wurde.[4] Dies ist teuer und zeitaufwendig. In der Pflanzenzüchtung löst man das Problem durch die Erzeugung ingezüchteter Linien. In letzter Konsequenz sind die homologen Chromosomen eines Individuums aus solch einer Linie identisch (nur homozygote SNPs in einem Individuum). Die Bestimmung der Haplotypen reduziert sich auf 1. und somit auf eine einmalige Sequenzierung eines Chromosoms bzw. Locus.
- ii) Bioinformatisch:
- Nicht immer ist das Geld und/oder Zeit vorhanden Mehrfachsequenzierungen durchzuführen und/oder die Möglichkeit gegeben Inzuchtlinien zu erzeugen. Tauchen nun bei Einmalsequenzierung heterozygote SNPs in einem Individuum auf und wird mehr als ein SNP betrachtet so können sich verschiedene mögliche Haplotypen für ein Individuum ergeben. Um aus diesen exponentiell vielen Möglichkeiten (bei linear anwachsender Anzahl an SNPs) eine biologisch sinnvolle auszuwählen wurden verschiedene Methoden, basierend auf unterschiedlichen Annahmen, entwickelt:
- ii.1) Basierend auf einem Sparsamkeitskriterium[5] (Parsimony based, siehe auch Ockham's Razor). Diese Methode versucht die Anzahl der Haplotypen zu minimieren welche benötigt werden um die SNPs einer gegebenen Population zu erklären. Es gibt verschiedene Ansätze basierend auf SAT[3][4] oder Linearer Programmierung[6] dieses Problem effizient zu lösen.
Weitere Eigenschaften: Wird unter der Annahme angewendet dass im betrachteten Locus keine oder kaum Rekombination stattfindet. Eine gefundene Lösung ist im Sinne des Sparsamkeitskriterium immer optimal. Nicht praktikabel für large-scale Analysen. - ii.2) Maximum-likelihood (mit Hilfe von Expectation-Maximization-Algorithmus[7] oder Monte-Carlo-Simulation[8]). Diese Methoden versuchen den Satz an Haplotypen zu finden (und die entsprechende Aufteilung auf die einzelnen Individuen) so dass die durch eine gegebene Zielfunktion berechnete Wahrscheinlichkeit der beobachteten Daten maximiert wird.
Weitere Eigenschaften: Anwendbar auch bei Rekombination.[9] Lösungen sind meist nur suboptimal, da Algorithmus in lokalem Optimum endet bzw. Vereinfachungen vornehmen muss damit eine Lösung überhaupt berechenbar ist. Praktikabel für large-scale Analysen wenn auch suboptimale Lösung ausreichend ist.
- ii.1) Basierend auf einem Sparsamkeitskriterium[5] (Parsimony based, siehe auch Ockham's Razor). Diese Methode versucht die Anzahl der Haplotypen zu minimieren welche benötigt werden um die SNPs einer gegebenen Population zu erklären. Es gibt verschiedene Ansätze basierend auf SAT[3][4] oder Linearer Programmierung[6] dieses Problem effizient zu lösen.
- Für beide Methoden ist eine Population mit mehr als einem Individuum notwendig damit die Grundannahmen greifen und biologisch sinnvolle Aussagen gemacht werden können. Teilprobleme des Haplotypenproblems sind NP-vollständig da sie durch SAT darstellbar sind (Satz von Cook) und im schlechtesten Fall dieselbe Komplexität wie SAT aufweisen, das Gesamtproblem ist somit NP-schwer.[3]
Quellen
Einzelnachweise
- ↑ The International HapMap Consortium (2003): The International HapMap Project. Nature 426, S. 789-796 [1]
- ↑ Ceppellini, R., E. S. Curtoni, P. L. Mattiuz, V. Miggiano, G. Scudeller and A. Serra: Genetics of leucocyte antigens. A family study of segregation and linkage. Histocompatibility Testing 1967, S. 149–185
- ↑ a b c d Efficient haplotype inference with Boolean Satisfiability. Lynce I, Marques-Silva JP. National Conference on Artificial Intelligence (AAAI) 2006. [2]
- ↑ a b c d e Haplotype inference from unphased SNP data in heterozygous polyploids based on SAT. Neigenfind J, Gyetvai G, Basekow R, Diehl S, Achenbach U, Gebhardt C, Selbig J, Kersten B. BMC Genomics 2008, 9:356. [3]
- ↑ Haplotype inference by Pure Parsimony. Gusfield D. Proceedings of the 14th annual Symposium on Combinatorial Pattern Matching 2003, 144-155. [4]
- ↑ A new Integer Programming formulation for the Pure Parsimony Problem in haplotype analysis. Brown DG, Harrower IM. WABI 2004, 254-265. [5]
- ↑ Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Excoffier L, Slatkin M. Mol Biol Evol 1995, 12:921–927. [6]
- ↑ Bayesian Haplotype Inference for Multiple Linked Single-Nucleotide Polymorphisms. Niu T, Qin ZS, Xu X, Liu JS. Am J Hum Genet 2002, 70(1): 157–169. [7]
- ↑ Inference of haplotypic phase and missing genotypes in polyploid organisms and variable copy number genomic regions. Su S, White J, Balding D, Coin L. BMC Bioinformatics 2008, 9:513. [8]
Literatur
- Lexikon der Biologie. 7. Band, Spektrum Akademischer Verlag, Heidelberg, 2004. ISBN 3-8274-0332-4
- Lewin, Benjamin: Molekularbiologie der Gene. Spektrum Akademischer Verlag, Heidelberg, Berlin, 1998. ISBN 3-8274-0234-4
Weblinks
Wikimedia Foundation.