- Hippel-Lindau Krankheit
-
Klassifikation nach ICD-10 Q85.8 Sonstige Phakomatosen ICD-10 online (WHO-Version 2006) 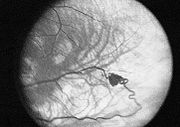 Hämangioblastom der Netzhautgefäße
Hämangioblastom der NetzhautgefäßeDas von Hippel-Lindau-Syndrom (VHL), gelegentlich auch als Retino-cerebelläre Angiomatose bezeichnet, ist eine seltene, erbliche Tumorerkrankung aus dem Formenkreis der sogenannten Phakomatosen. Die Patienten entwickeln gutartige, geschwulstähnliche Gewebsveränderungen (Angiome) vornehmlich im Bereich der Netzhaut des Auges und des Kleinhirns. Im zentralen Nervensystem können darüber hinaus auch der Hirnstamm und das Rückenmark, selten Großhirn betroffen sein. Charakteristisch für das VHL-Syndrom ist es, dass sich aus Vorformen des Bindegewebes Geschwulste entwickeln, die aus Gefäßknäueln bestehen. Viele Patienten haben auch Gewebsveränderungen im Bereich von Niere (Nierenzellkarzinome), Nebenniere (Phäochromozytom) und Bauchspeicheldrüse. Bei Männern kann der Nebenhoden betroffen sein. Diese Gewebsveränderungen können harmlos sein, sich aber auch zu bösartigen Tumoren entwickeln. Die Ursache der Erkrankung ist eine Genmutation. Da es sich bei dem VHL-Syndrom um eine genetische Erkrankung handelt, ist keine Heilung möglich. Die Behandlung der Patienten richtet sich nach dem Ort und der Ausprägung der Gewebsveränderungen. Am Auge werden die Netzhautgeschwulste mittels Laserstrahlen zerstört. Bösartige Geschwulste treten vor allem im Bereich der Niere auf und werden gemäß den Richtlinien der Behandlung dieser Erkrankung behandelt. Die Gewebsveränderungen im zentralen Nervensystem werden operiert, wenn durch deren Lage und Größe gefährliche Folgen für die Patienten eintreten können. Da die Krankheit schon frühzeitig erkannt werden kann, werden regelmäßige Kontrolluntersuchungen empfohlen.
Inhaltsverzeichnis
Geschichte
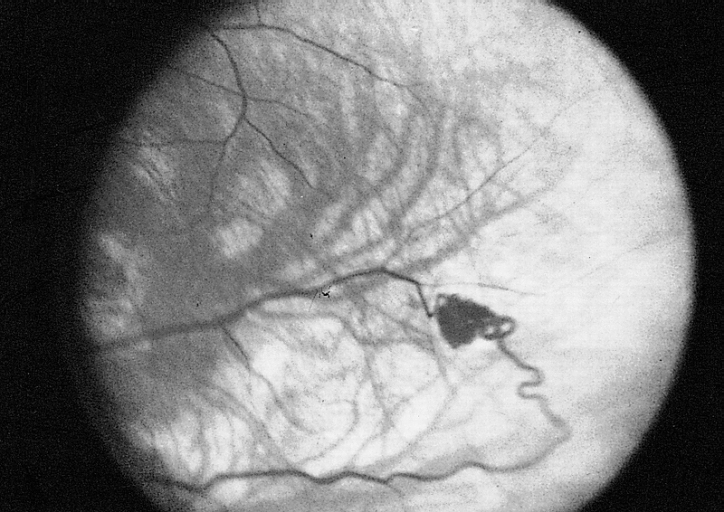
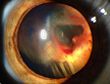 Netzhautveränderungen bei VHL
Netzhautveränderungen bei VHLNamensgeber der Erkrankung sind der Göttinger Ophthalmologe Eugen von Hippel (1867-1939) und der schwedische Pathologe Arvid Lindau (1892-1958). Von Hippel beschrieb 1904 erstmalig Angiome des Auges, Lindau 1926 die Angiome im Rückenmark.
Inzidenz, Erbgang, Epidemiologie
Das VHL-Syndrom ist eine seltene autosomal-dominante Tumorerkrankung. Die Inzidenz wird zwischen 1:36.000 - 1:45.000 angegeben. Je nach Studie beträgt die Spontanmutationsrate bis zu 50 %. Männer wie Frauen sind gleichermaßen betroffen.
Pathogenese, Molekularbiologie und pathophysiologische Zusammenhänge
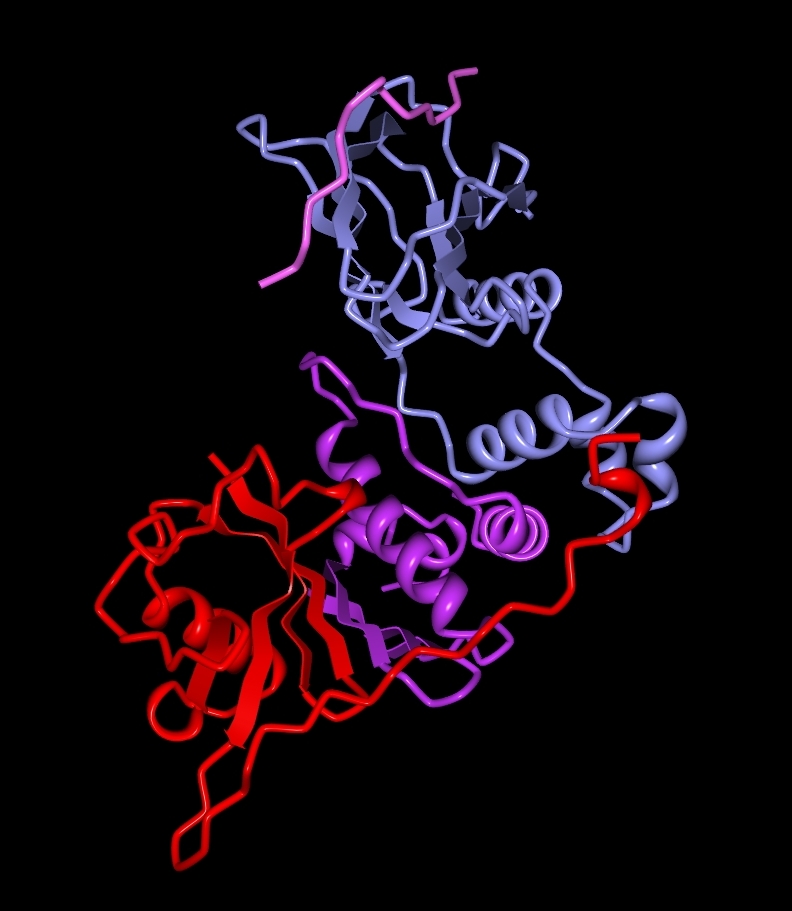
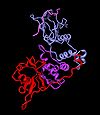 VHL-Komplex
VHL-KomplexDas Gen für die Hippel-Lindau Erkrankung wurde im Bereich von Chromosom 3, Bande p25/26 lokalisiert. Es ist im Zellzyklus und der Gefäßneubildung involviert. Das HL-Gen besitzt drei Exons und kodiert für ein nukleäres Protein, das eine Bindung mit Proteinen der Elongin-Gruppe eingeht. Bei Patienten mit einer HL-Erkrankung wurde eine Vielzahl von Mutationen entdeckt, die alle weitgehend gleichmäßig über das Gen verteilt sind. In verschiedenen Studien wurde festgestellt, dass 35 % der Mutationen Missense-Mutationen sind und etwa 75 % der Patienten haben eine Keimbahnmutation.
Pathologie
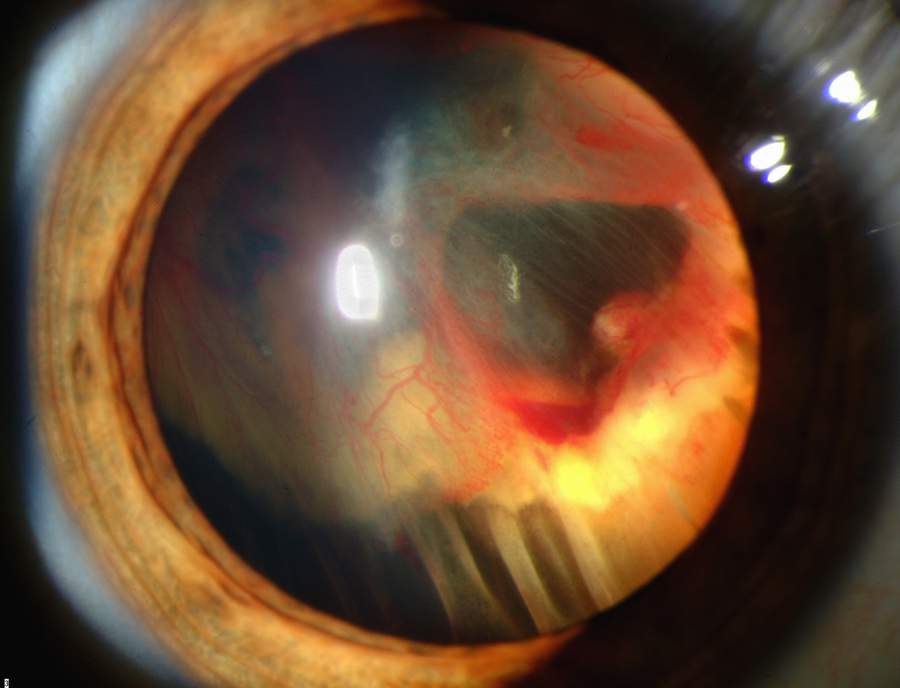
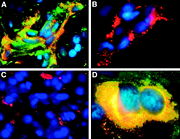 Hämangioblastom
HämangioblastomBei den Angiomen des HL-Syndroms handelt es sich vorwiegend um kapilläre Hämangiome und Hämangioblastome. Als Hämangiom bezeichnet man gutartige, geschwulstähnliche Neubildungen mit einer Ausprägung als Blutgefäßknäuel. Sie treten meist als Hamartome auf, sind also keine Tumoren im engeren Sinne. Die Hämangiome entstehen nicht, wie der Name nahe legt aus Blutgefäßen, sondern aus Bindegewebsvorläufern und sie entwickeln sich zu Strukturen, die man am einfachsten als Blutgefäßknäuel oder Blutschwämme bezeichnen kann. Die Blutschwämme, die sich im Kleinhirn, Hirnstamm und Rückenmark der VHL-Patienten finden sind Hämangioblastome. Mit diesem Begriff bezeichnet man echte Neubildungen, die aus gewucherten Kapillarsprossen bestehen.
- Die retinalen Hämangiome können eine mehr angiomatöse oder mehr fibrosierende Ausprägung haben. Dies ist für die Prognose der Erkrankung in Bezug auf das Auge sehr wichtig. Bei mehr angiomatösen Hämangiomen überwiegt der Gefäßanteil der Gewebsveränderung, was häufiger zu massiven Einblutungen in das Auge mit einer plötzlichen vollständigen Erblindung führen kann. Fibrosierende Hämangiome machen eher eine Traktionsveränderung der Netzhaut. Wenn die Hämangiome in der Peripherie der Netzhaut gelegen sind, machen sie häufig keinerlei Beschwerden. Liegen sie dagegen zentral, kann bald eine Visusminderung auftreten. Wenn die Gefäßknäuel Kurzschlüsse zwischen Venen und Arterien ausbilden, kann es zum Austreten von Gewebsflüssigkeit im Auge kommen mit entsprechenden Folgen (Druckerhöhung).
- Hämangioblastome manifestieren sich bevorzugt im Kleinhirn und werden bei langsamen Wachstum durch Störung des Liquorabflusses symptomatisch. Da sie auch Erythropoietin ausbilden, wird in manchen Fällen eine Polyzythämie beobachtet. Unter dem Mikroskop stellt sich das kapilläre Hämangioblastom als äußerst gut vaskularisierter Tumor dar mit CD31/CD34-positiven Gefäßendothel und NSE-Expression des Stromas dar.
Krankheitsbild
 Pankreaskopfzyste
PankreaskopfzysteDie Kern- oder Kardinalsymptome des VHL-Syndroms sind das Auftreten von Hämangiomen der Retina und Hämangioblastomen des Kleinhirns.
Das klinische Spektrum der Erkrankung umfasst neben den Affektionen von Augen und Kleinhirn das Auftreten von Hämangioblastomen im Bereich des Hirnstamms und des Rückenmarkes. Sodann werden Nierenzellkarzinome (Erkrankungsrisiko liegt bei 25 - 45 %, meist ab dem 50. Lebensjahr), Pankreaszysten, Phäochromozytome, Nebenhodenzysten und eine Polyzythämie beobachtet.
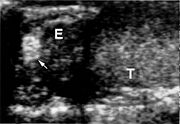 Nebenhodenzyste
NebenhodenzysteDie Hämangioblastome des ZNS präsentieren sich in ca 60 % der Fälle im Kleinhirn, in 13 % der Fälle im Rückenmark und in 4 % der Fälle im Hirnstamm. Selten < 1% im Großhirn. Die Läsionen im Kleinhirn werden im Schnitt bei Patienten im Alter von 29 Jahren klinisch auffällig und im Fall von Rückenmarksläsionen im Alter von 34 Jahren.
Als diagnostisches Kriterium gilt der Nachweis von beidseitigen oder multiplen retinalen Hämangiomen oder der Nachweis multipler Hämangioblastome der hinteren Schädelgrube.
Aufgrund klinischer Verläufe und Unterschieden in der Phänotyp-Genotyp-Korrelation unterscheidet man zwei Formen des VHL-Syndroms. Patienten mit einem Phäochromozytom ordnet man dem VHL Typ I zu, Patienten ohne Phäochromozytom ordnet man dem VHL-Typ II zu.
Zum Verlauf der Erkrankung werden sehr unterschiedliche Angaben gemacht. Die retinalen Angiome werden im Durchschnitt bei den Patienten im Alter von 25 aufgedeckt. Die Netzhautveränderungen können, sofern sie rechtzeitig entdeckt werden gut behandelt werden. Die intrazerebralen und spinalen Hämangioblastome können vor allem im Bereich des Hirnstammes zu gefährlichen Blutungen führen.
Genotyp-Phänotyp-Korrelation
Bei dem HL-Typ I wurden in über 50 % der untersuchten Fälle Mikrodeletionen und Nonsensemutationen entdeckt. Beim HL-Typ II fanden sich in fast 100 % der untersuchten Patienten Missense-Mutationen.
Diagnose
Die Diagnose VHL-Syndrom wird beim Vorhandensein von kapillären Hämangioblastomen (gefäßreichen Tumoren) im ZNS oder der Netzhaut des Auges gestellt. Weitere zum VHL-Komplex gehörende Tumoren (Phäochromozytom, Nierenzellkarzinom) oder eine entsprechende Familienanamnese treten hinzu. In der Kernspintomografie stellen sich die Hämangioblastome als Kontrastmittelaufnehmende Knötchen dar.
Therapie
Therapie ist die chirurgische Entfernung der Tumore. Alternative Behandlungsmethoden wie Bestrahlung, z.B. mittels Gamma-Knife oder Linearbeschleuniger sind ebenfalls möglich.
Quellen
Bücher
- Olaf Rieß und Ludger Schöls (Hrsg.): Neurogenetik. Molekulargenetische Diagnostik neurologischer Erkrankungen. Springer. Berlin 1998. ISBN 3-540-63874-1.
- Lewis P. Rowland (Ed.): Merrits Textbook of Neurology. Williams and Wilkins. Baltimore 1995. ISBN 0-683-07400-8.
- Mark S. Greenberg: Handbook of Neurosurgery. Lakeland 1997. ISBN 0-9626384-5-5.
- Bruce O. Berg (Ed.): Principles of Child Neurology. McGraw-Hill. New York 1996. ISBN 0-07-005193-3.
- Raymond D. Adams (Ed.): Principles of Neurology. McGraw-Hill. New York 1997. ISBN 0-07-067439-6.
Fachartikel
- Constans JP et. al. (1986): Posterior fossa hemangioblastomas. Surg. Neurol. 25:269-275. PMID 3945908
- Hough DM et. al. (1994): Pancreatic lesions in von Hippel-Lindau diseas: Prevalence, clinical significance and CT findings. AM J Radiol 162:1091-1094. PMID 8165988
- Maher ER et. al. (1991): Familial renal cell carcinoma. Clinical and molecular genetic aspects. Brit J Cancer 63:176-179. PMID 1997093
- Maher ER et. al. (1991): Von Hippel-Lindau disease: a genetic study. J Med Genet 28:443-447. PMID 1895313
- Melmon KL et al. (1964): Lindaus disease: Review of the literature and study of a large kindred. Am J Med 36:595-617. PMID 14142412
- Neumann HPH et. al. (1991): Clustering of features of Hippel-Lindau syndrom. Lancet 337: 1062-1054. PMID 1673491
- Neumann HPH et. al. (1995): Von Hippel Lindau Syndrom. Brain Pathol 5:181-193. PMID 7670659
- Whaley JM et. al. (1994): Germ-line mutations in the von Hippel-Lindau tumor-suppressor gene are similar to somatic von Hippel-Lindau aberrations in sporadic renal cell carcinoma. Am J Hum Genet. 1994 Dec;55(6):1092-102. PMID 7977367

Bitte beachte den Hinweis zu Gesundheitsthemen!

Wikimedia Foundation.