- IR-Spektrometer
-
Infrarotspektroskopie, oft auch nur IR-Spektroskopie genannt, ist ein physikalisches Analyseverfahren, das mit infrarotem Licht (800–500.000 nm) arbeitet. Das Verfahren gehört zu den Methoden der Molekülspektroskopie, die auf der Anregung von Energiezuständen in Molekülen beruhen. Die IR-Spektroskopie wird zur quantitativen Bestimmung von bekannten Substanzen, deren Identifikation anhand eines Referenzspektrums oder zur Strukturaufklärung unbekannter Substanzen genutzt. Die dabei hauptsächlich eingesetzte Messtechnik ist die Fourier-Transformations-IR-Spektroskopie. Ähnliche molekülspektroskopische Methoden sind die Ramanspektroskopie, die ebenfalls im Infrarotbereich, und die UV/VIS-Spektroskopie im höherliegenden Frequenzbereich.
Inhaltsverzeichnis
- 1 Varianten
- 2 Messprinzip
- 3 IR-Spektroskopie-Techniken
- 4 Siehe auch
- 5 Einzelnachweise
- 6 Literatur
- 7 Weblinks
Varianten
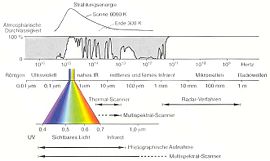 Elektromagnetisches Spektrum
Elektromagnetisches SpektrumAus spektroskopischer Sicht wird zwischen dem nahen Infrarot (NIR; Wellenzahl: 12500–4000 cm−1; Wellenlänge: 0,8–2,5 µm, vgl. Nahinfrarotspektroskopie), dem mittleren oder klassischen (normalen) Infrarot (MIR; Wellenzahl: 4000–400 cm−1 Wellenlänge: 2,5–25 µm) und dem fernen Infrarot (FIR; Wellenzahl: 400–10 cm−1; Wellenlänge: 25–1000 µm) unterschieden, da durch die Absorption im jeweiligen Bereich unterschiedliche Phänomene zu beobachten sind. Eine Absorption im FIR-Bereich führt überwiegend zur Rotation ganzer Moleküle. Im MIR-Bereich und NIR-Bereich wird die Schwingung von Atomen bzw. Atomgruppen an ihren Molekülbindungen angeregt, wobei im NIR-Bereich die sogenannten Oberschwingungen des MIR-Bereich detektierbar sind (insbesondere von CH-, OH- und NH-Bindungen). [1]
Die Spektroskopie im mittleren Infrarot – häufig nur als IR-Spektroskopie bezeichnet – ist eine leistungsfähige Technik in der chemischen Analytik organischer Substanzen. Sie ermöglicht direkte Aussagen über Vorhandensein und ggf. Konzentration infrarot-aktiver funktioneller Gruppen. Die Nahinfrarotspektroskopie (NIRS) wird zur schnellen Überprüfung von Stoffen und Stoffgemischen genutzt, ist aber auf computergestützte chemometrische Modelle mit begleitender, stichprobenhafter Referenzanalytik angewiesen.
In IR-Spektren wird, im Gegensatz zu UV-Spektren, häufig nicht die Absorption sondern die Transmission als Maß für die Durchlässigkeit der Anregungsstrahlung verwendet. Die Transmission wird nach oben zunehmend der vertikalen Achse aufgetragen – Bereiche geringer Durchlässigkeit der IR-Strahlung ergeben einen Ausschlag nach unten. Die Darstellung als Absorptionspektrum wird aber, vor allem bei der ATR-Technik, ebenfalls genutzt.
Messprinzip
Absorption von IR-Strahlung
Bei der Bestrahlung eines Stoffes mit elektromagnetischen Wellen werden bestimmte Frequenzbereiche absorbiert. Infrarotstrahlung liegt energetisch im Bereich der Rotationsniveaus von kleinen Molekülen und den Schwingungsniveaus von Molekülbindungen, d. h., die Absorption führt zu einer Schwingungsanregung der Bindungen. Sie sind in Form von Ausschlägen im gemessenen Spektrum (Diagramm) sichtbar. Da die dazu notwendigen Energien bzw. Frequenzen charakteristisch für die jeweiligen Bindungen sind, können so auch Materialien identifiziert werden. Die IR-Spektroskopie ist somit strukturaufklärend.
Wechselwirkung zwischen elektromagnetischer Strahlung und dem Molekül kann nur dann auftreten, wenn im Molekül bewegte elektrische Ladung zur Verfügung steht. Das ist immer dann der Fall, wenn das Molekül entweder ein veränderbares oder ein induzierbares Dipolmoment aufweist (IR-aktiv). In Molekülen mit Schwingungen symmetrisch zum Symmetriezentrum treten keine Änderungen des Dipolmoments auf (IR-inaktiv). Solche "verbotenen" Schwingungen sind allerdings oft raman-aktiv.
Der einfachste Fall ist ein zweiatomiges Molekül. Bei mehratomigen Molekülen kommt es zur Überlagerung von Grundschwingungen. Dementsprechend sieht man eine Reihe von Absorptionsbanden, die interpretiert werden müssen.
Mechanisches Modell
 Molekül zwischen zwei ungeladenen Kondensatorplatten
Molekül zwischen zwei ungeladenen Kondensatorplatten Molekül richtet sich im elektromagnetischen Feld aus und vergrößert seinen Bindungsabstand
Molekül richtet sich im elektromagnetischen Feld aus und vergrößert seinen BindungsabstandDas denkbar einfachste Modell, welches zur Erklärung von Vibrations- und Rotationsanregungen verwendet werden kann, ist das klassische Modell der Wechselwirkung eines permanenten elektrischen Dipols im elektromagnetischen Feld; Später können in besseren Modellen, wie dem quantenmechanischen Modell, auch Moleküle ohne ein permanentes Dipolmoment beschrieben werden.
Zwischen den Atomen und ihren Nachbarn bestehen anziehende und abstoßende Wechselwirkungen. Daher befindet sich der optimale Bindungsabstand im Molekül im Minimum der Potentialfunktion. Mechanistisch lässt sich dies so vorstellen, als wären die Atome durch Federn miteinander verbunden. Die Kraft, die man zur Auslenkung einer Feder braucht, wird mit dem hookeschen Federgesetz beschrieben. Bringt man nun ein solches Molekül in ein elektrisches Feld, wie es beispielsweise in einem Plattenkondensator besteht, wird sich das Molekül erstens mit seinem Dipolmoment entlang des elektrischen Feldes ausrichten und zweitens seinen Bindungsabstand vergrößern. Wird jetzt Wechselspannung angelegt oder regt man das Molekül mit einer elektromagnetischen Welle an, fangen die an den Bindungen „hängenden“ funktionellen Gruppen zu schwingen und zu rotieren an. Das mechanistische Modell ist allerdings nur sehr begrenzt tauglich, da es beispielsweise nicht erklärt, warum nur diskrete Energien zur IR-Anregung zugelassen sind und warum auch Moleküle ohne permanentes Dipolmoment eine IR-Absorption zeigen.
Quantenmechanisches Modell
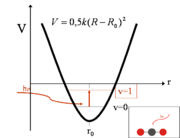 Parabel des quantenmechanischen Modells
Parabel des quantenmechanischen ModellsWie auch im Modell der klassischen Mechanik ist die Grundlage des quantenmechanischen Modells der Vibrations- und Rotationsanregung die Potentialfunktion. Die Potentialfunktion lässt sich in ihrem Minimum gut durch eine Parabel annähern. Eine solche Parabel ergibt sich aus der Integration des hookschen Federgesetzes.
Wird jetzt das Molekül mit elektromagnetischer Strahlung angeregt, kann das Molekül anfangen zu schwingen, wenn die Energie ausreicht, es aus seinem Grundzustand in den ersten angeregten Schwingungszustand zu heben. Um diese Energie zu bestimmen, muss die Schrödinger-Gleichung für dieses Potential gelöst werden. Nach Abtrennung der Relativbewegung von Atomkernen und Elektronen ergibt sich als Lösung der Schrödinger-Gleichung ein Zusammenhang zwischen der benötigten Energie, Bindungsstärke (k) und der reduzierten Masse (μ). Anders als beim klassischen harmonischen Oszillator ist im Quantenmechanischen Fall die Schwingungsenergie durch die Schwingungsquantenzahl v gequantelt.
Schrödinger-Gleichung:
Lösung der Schrödinger-Gleichung:
Schwingungsformen
In anorganischen und organischen Substanzen treten bei Absorption von Strahlung aus dem infraroten Bereich mechanische Schwingungen auf. Es können drei verschiedene Arten von Schwingungen unterschieden werden:
- Valenzschwingungen (Streckschwingungen): Schwingungen entlang der Bindungsachse zweier Atome oder Molekülteile durch eine Dehnung oder Stauchung der Bindung
- Deformationsschwingungen in der Ebene (Biege-/Beugeschwingungen): Schwingungen unter der Deformation des Bindungswinkels
- Deformationsschwingungen außerhalb der Ebene (Dreh-/Kippschwingungen): Schwingungen unter der Deformation des Bindungswinkels senkrecht zur Bindungsebene
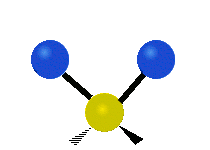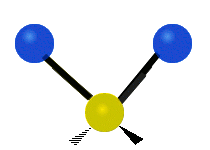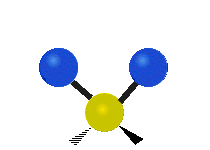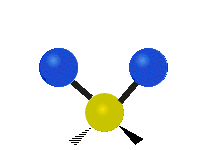
Symmetrische Streckschwingung
(engl. stretching)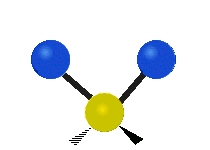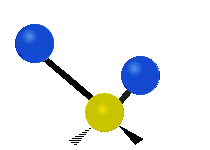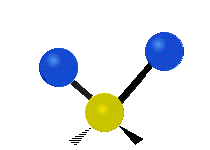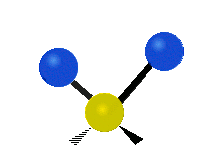
Asymmetrische Streckschwingung
(engl. stretching)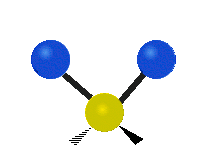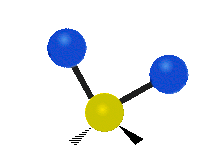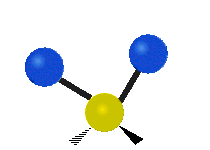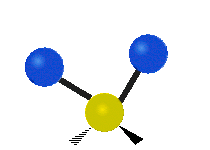
„Schaukelschwingung“
(engl. rocking)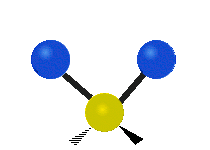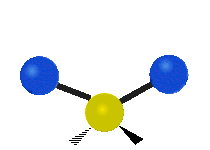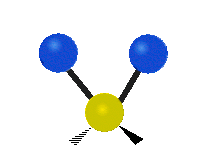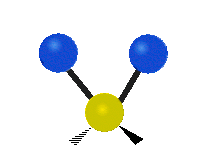
Scher-/Deformations-
schwingung
(engl. scissoring oder bending)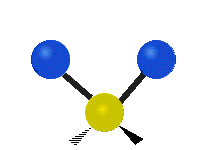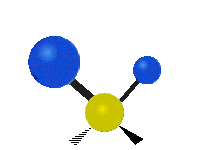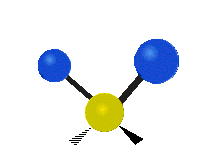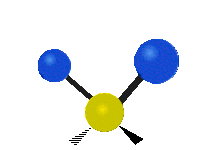
Drehschwingung
(engl. twisting oder torsing)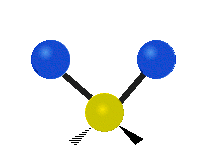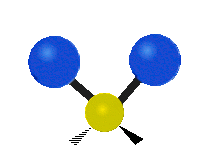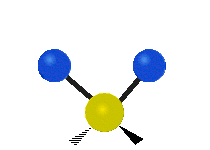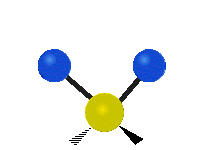
„Wippschwingung“
(engl. wagging)Da Molekülschwingungen bestimmter Atomgruppen im Bereich von 4000–1500 cm−1 besonders charakteristisch sind, eignet sich die IR-Spektroskopie zur Bestimmung der funktionellen Gruppen des untersuchten Moleküls. Zusätzlich ist das gesamte Spektrum und besonders der Fingerprintbereich, im Bereich von 1500–600 cm−1, zwar nicht für buchstäblich jede Substanz charakteristisch, wie oft behauptet, bietet aber oft wertvolle Anhaltspunkte zur Identifikation. Enantiomere chiraler Verbindungen zeigen nicht grundsätzlich unterschiedliche Banden im Fingerprintbereich und keinesfalls in irgendeinem anderen.
Rotations-Schwingungs-Spektrum
Das ideale Rotations-Schwingungs-Spektrum
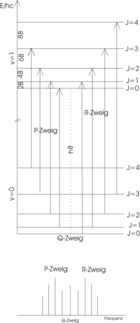 Das ideale Rotations-Schwingungs-Spektrum von Chlorwasserstoff
Das ideale Rotations-Schwingungs-Spektrum von ChlorwasserstoffChlorwasserstoff (HCl) ist ein zweiatomiges Molekül mit einem ausgeprägten Dipolmoment. Das HCl-Molekül kann vereinfacht als ein linearer Kreisel angesehen werden. Die Quantenmechanik liefert für ein lineares Molekül die spezielle Auswahlregel für einen Rotationsübergang:
- ΔJ = + 1 oder ΔJ = − 1
wobei J die Rotationsquantenzahl darstellt. Wenn das Dipolmoment parallel zur Hauptrotationsachse liegt (wie z. B. beim Ammoniak), wäre auch ΔJ = 0 möglich. Für einen Schwingungsübergang des harmonischen Oszillators lautet die spezielle Auswahlregel:
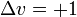 oder
oder 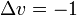
Die Übergänge zwischen den verschiedenen Rotationsniveaus (J = 0, 1, 2, …) des Schwingungsgrundzustandes (v = 0) und den Rotationsniveaus (J' = 0, 1, 2, …) des angeregten Schwingungszustandes (v' = 1) können in zwei Gruppen, den R-Zweig (rechts) für Übergänge mit ΔJ = +1 und den P-Zweig (links) für Übergänge mit ΔJ = −1, unterteilt werden und sind in der Abbildung skizziert. Da ΔJ = 0 für HCl nicht erlaubt ist, erscheint auch kein Q-Zweig. Im oberen Bild ist das Energieniveauschema abgebildet. Anhand der Pfeillänge ist deutlich zu erkennen, dass jeder Übergang eine Energiezufuhr von
erfordert, wobei
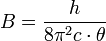 die Rotationskonstante ist. Diese beinhaltet das Trägheitsmoment
die Rotationskonstante ist. Diese beinhaltet das Trägheitsmoment 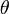 des Moleküls bezüglich der Rotationsachse.
des Moleküls bezüglich der Rotationsachse.Für die Energiedifferenz zum benachbarten Übergang gilt damit
Im idealen Rotationsschwingungsspektrum (unteres Bild) haben daher die Peaks einen konstanten Abstand von 2B. Im Experiment weisen die Peaks etwas andere Abstände auf, da das Modell nicht:
- die Anharmonizität der Schwingung
- den gedehnten Atomabstand durch die Zentrifugalkräfte (Näherung: starrer Rotator)
- und die Wechselwirkung von Schwingungs- und Rotationsbewegung beachtet.
Das reale Rotations-Schwingungs-Spektrum
 Rotations-Schwingungs-Spektrum von gasförmigem Chlorwasserstoff bei Raumtemperatur.
Rotations-Schwingungs-Spektrum von gasförmigem Chlorwasserstoff bei Raumtemperatur.Das reale Rotations-Schwingungs-Spektrum weicht von dem idealen ab aufgrund der Einschränkungen der Messmethoden und aufgrund von Kopplungen. Die Abstände der Peaks (Amplitudenausschlag im Diagramm, auch Banden genannt) betragen nicht wie im idealen Fall genau 2B, sondern werden mit steigender Wellenzahl kleiner, sowohl im P-Zweig als auch im R-Zweig. Das nebenstehende Diagramm zeigt ein aufgenommenes Spektrum von Chlorwasserstoff. Die Feinstruktur des Spektrums ist gut für beide Isotope des Chlors zu erkennen
Lage der IR-Absorptionsbanden
Stärkere chemische Bindungen und Atome kleinerer Masse verursachen im IR-Spektrum Absorptionsmaxima bei großen Wellenzahlen (hohe Energie), große Massen hingegen verursachen IR Absorptionsmaxima bei kleinen Wellenzahlen (niedrige Energie) (siehe Deuterierung). Die Energie ist proportional zum Quadrat des permanenten Dipolmomentes. Daher liefern polare Moleküle intensive Rotationsübergänge. Vergleicht man allerdings die Amplituden der Peaks eines einzelnen Moleküls miteinander, fällt auf, dass die Stärke der Übergänge mit wachsenden J zunächst schnell zunehmen, durch ein Maximum gehen und schließlich für große J wieder abfallen. Die Ursache dafür ist, dass die Stärke die Entartungen der verschiedenen Rotationszustände und die Besetzungszahlen der Rotationsniveaus im Ausgangszustand widerspiegelt. Der Grad der Entartung nimmt mit steigendem J zu, was zu einer höheren Energie führt. Andererseits nehmen die Besetzungszahlen mit steigender Energie ab, was schließlich zu einer Abnahme der Strahlungsstärke führt.
IR-Spektren werden dahingehend interpretiert, dass man aus der Kurve des gemessenen IR-Spektrums die Molekülgestalt herauszufinden versucht. Dabei misst man die verschiedenen Schwingungsvarianten der Moleküle. Ein typisches IR-Spektrum reicht von 4000 cm−1 bis 400 cm−1 (Wellenzahl). Die Wellenzahl wird in der etwas ungewöhnlichen Einheit cm−1 angegeben. Man kann aus der Wellenzahl allerdings bequem die Anregungsenergie berechnen, indem man die Wellenzahl mit dem planckschen Wirkungsquantum h und der Lichtgeschwindigkeit c multipliziert. Bei einem IR-Spektrum hinterlässt jedes Molekül ein typisches Muster. Ab etwa 1500 cm−1 abwärts lassen sich die Ursachen für das Spektrum kaum mehr auf bestimmte Molekülgruppen zurückführen. Dies ist der sogenannte Fingerprint-Bereich. Mithilfe dieses "Fingerabdrucks" lässt sich bei Reinsubstanzen ein Molekül eindeutig identifizieren, sofern ein Referenzspektrum vorliegt. Da in der Praxis allerdings meist keine Reinsubstanzen, sondern Gemische vorliegen, kann aus einem IR-Spektrum oft keine exakte Zusammensetzung hergeleitet werden. Meist genügt es schon zu wissen, welche Stoffgruppen mithilfe des IR-Spektrums entdeckt worden sind.
Schwingungsdaten von wichtigen Molekülgruppen
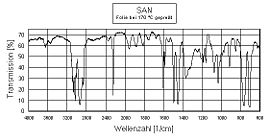 Transmissionsinfrarotspektrumm eines Styrol-Acrylnitril-Copolymers (SAN)
Transmissionsinfrarotspektrumm eines Styrol-Acrylnitril-Copolymers (SAN)Die folgende Auflistung enthält exemplarisch einige Molekülgruppen und die zugehörigen Bereiche der Absorptionsbanden:
Bezeichnung/ Molekülgruppe Wellenzahlbereich
in cm−1Bemerkung ν(–C–Hx) 2850–3200 ν = Streckschwingung; x = 1…3, Anzahl der gebundenen Wasserstoffatome δ(–C–Hx) 1400 δ = Deformationsschwingung; x = 1…3, Anzahl der gebundenen Wasserstoffatome –C=C 1650 –C≡C 2200–2500 ν(–OH) 3200–3600 sehr typisch: verschmiert über große Breite ν(–OH) 2500–3000 in Carboxylen –C=O 1700 ν(–C≡N) 2200–2260 Valenzschwingung, z. B. in ABS oder SAN (s. Bild) ν(–NH) 3100–3500 –NO2 1500 –C–X < 1500 wobei X für ein Halogen steht O=C=O 2349 Kohlendioxid, erscheint in Spektren infolge der unterschiedlichen Weglängen der beiden Lichtstrahlen (dispersive Geräte) oder nicht ausreichender Spülung mit Stickstoff (FT-IR-Geräte). IR-Spektroskopie-Techniken
Transmission
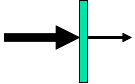 Prinzip der Transmissionsmessung
Prinzip der TransmissionsmessungEine der am häufigsten eingesetzten Methoden ist die Messung des Transmissionsgrades einer Probe. Dazu wird eine Probe mit Infrarot durchstrahlt und der Anteil der Messstrahlung gemessen, der die Probe durchdrungen hat. Besitzt die Probe IR-aktive Bindungen oder Molekülgruppen sind im Transmissionsspektrum Bereiche geringerer Intensität (Absorptionsbanden) zu sehen. Die Intensität der Bande nimmt gemäß dem Lambert-Beerschen Gesetz (unter Anderem) mit der Probendicke zu. Zusätzlich wird auch noch ein Teil der Strahlung reflektiert], entsprechende Anteile wirken sich meist als über das gesamte Spektrum erhöhte verringerte Transmission aus und können mit einer Grundlinienkorrektur großteils entfernt werden.
Je nach Probenform (gasfömig, flüssig, fest) gibt es spezielle Messtechniken, die sich in der Art der Probvenvorbereitung und der Messzellen (Probenhalter mit mehr oder weniger komplizierten Strahlenwegen) unterscheiden. Die einfachste Messanordung entspricht einem simplen Probenhalter der einen Feststoff (beipielsweise eine Folie oder Platte) im Strahlengang des Spektrometers fixiert. Bei Proben mit parallelen Grenzflächen (wie bei einer Folie) entstehen jedoch Interferenzen durch Mehrfachreflexionen in der Probe (siehe auch Newtonsche Ringe) die je nach Dicke und Planparallelität das Spektrum verfälschen, die Interferenzen können aber auch für eine Schichtdickenbestimmung genutzt werden.
 Presswerkzeug zur Herstellung von KBr-Presslingen (Einzelteile)
Presswerkzeug zur Herstellung von KBr-Presslingen (Einzelteile) Presswerkzeug im Einsatz
Presswerkzeug im Einsatz Einfache Transmissionhalterung für die Infrarotspektroskopie von 13-mm-KBr-Presslingen. Anbei zwei Presslinge aus reinem KBr (rechts) und einer stark streuenden Probe in KBr (links, weißer Pressling)
Einfache Transmissionhalterung für die Infrarotspektroskopie von 13-mm-KBr-Presslingen. Anbei zwei Presslinge aus reinem KBr (rechts) und einer stark streuenden Probe in KBr (links, weißer Pressling)Eine weit verbreitete Technik feste Proben zu messen, ist die Einbettung der Festkörpers in ein IR-transparentes Trägermaterial (auch Matrix genannt). Man verwendet hauptsächlich anorganische Salze wie Alkalihalogenide, eine gängige Matrix ist Kaliumbromid (KBr). Zur Messung von Feststoffen wird trockenes Kaliumbromid-Pulver (KBr ist hygroskopisch) mit einem kleinen Anteil der zermahlenen Probensubstanz vermengt und anschließend unter hohem Druck gepresst. Bei diesem Druck wird das Kaliumbromid plastisch und es entstehen durch kalten Fluss (meist auch optisch) transparente „Presslinge“. Diese können dann einfach im Strahlengang platziert werden.
Flüssige Proben können zwischen zwei IR-transparente Platten platziert werden. Dies kann statisch oder auch dynamisch in Form einer Durchflusseinheit erfolgen. Gasförmige Proben werden in der Regel in spezielle Küvetten eingebracht.
Schichtdickenmessung
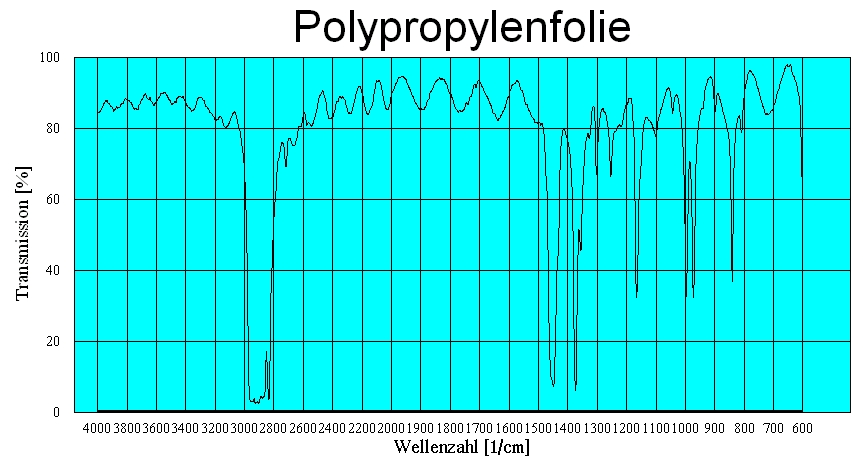
 Polypropylenfolie mit Interferenzen (Dicke ca. 38 µm)
Polypropylenfolie mit Interferenzen (Dicke ca. 38 µm) Polypropylenfolie ohne Interferenzen
Polypropylenfolie ohne InterferenzenDa es bei planparallelen dünnen Proben, z. B. Folien zu Interferenzen kommt, wird das eigentliche Spektrum von einem sinusförmigen Wellenzug überlagert. Aus der Lage der Maxima kann man die Dicke der Folie berechnen.
mit n = Anzahl der Maxima zwischen ν1 und ν2
Eliminieren kann man die Interferenzen bei Folien indem man sie knittert oder aufrauht.
Christiansen-Effekt
Der Christiansen-Effekt (nach Christian Christiansen) ist ein Artefakt bei IR-Transmissionsmessungen von in Kaliumbromid oder einer anderen Matrix eingebundenen Substanzen (so genannte Presslinge). Er äußert sich vor allem durch asymmetrische Banden im Spektrum. Dabei zeigt die hochfrequente Flanke einen sehr steilen Verlauf (oft höher als die Basis des Spektrums). Die niederfrequente Flanke zeigt dementgegen eher ein flachen, langgezogenen Verlauf. Die Ursache liegt in der anormalen Dispersion in der Nähe starker Absorptionsbanden. Beobachtet wird der Effekt jedoch nur bei Presslingen, bei denen die zu untersuchende Substanz zu grobkörnig ist, oder bei Proben mit größerem Unterschied im Brechungsindex zwischen der zu untersuchenden Substanz und dem Matrixmaterial. Das heißt, der Effekt kann minimiert bzw. verhindert werden, wenn die Substanz durch Zermahlen stärker verkleinert wird.[2][3][4]
Siehe auch: Christiansen-Filter
Reflexion und Absorption
 Prinzip der Reflexionsmessung
Prinzip der Reflexionsmessung Messeinrichtung für Reflexionsmessungen
Messeinrichtung für Reflexionsmessungen Im Strahlengang eingebaute Messeinrichtung für Reflexionsmessungen, an der durch das rote "X" gekennzeichneten Stelle wird die Probe aufgebracht
Im Strahlengang eingebaute Messeinrichtung für Reflexionsmessungen, an der durch das rote "X" gekennzeichneten Stelle wird die Probe aufgebrachtEine andere Methode ist die (externe) Reflexion der IR-Strahlung an einer glatten Oberfläche. Die Methode nutzt den Umstand, dass der Reflexionsgrad einer Probe abhängig vom komplexen Brechungsindex der Proben ist (vgl. Fresnelsche Formeln). Im Bereich der Schwingungszentren steigt die Absorption, was im komplexen Brechungsindex durch einen Ansteig des Extinktionskoeffizienten (der Imaginärteil des Brechungsindexes) dargestellt wird. Dies führt zu einer erhöhten Reflexion des Probe für diesen Frequenzbereich und einer Bande im Relfexionsspektrum.
Die Technik eignet sich unter anderem für die Messung stark absorbierender Proben. Hauptnachtteil der Methode ist, dass sich die Reflexionsspektren stark von Transmissionsspektren unterscheiden. Die Spektren zeigen derivationsähnliche Bandenformen, deren Ursache in der anomalen Dispersion in der Nähe von Absorptionszentren eines Materials, das heißt die die Brechzahl steigt hier mit der der Wellenlänge an, anstatt wie sonst zu fallen. Dies fürht zu einer geänderten Reflektivität und somit zu den beobachteten verzerrten Banden. Für reine Reflexionsspektren kann mit Hilfe der Kramers-Kronig-Transformation (KKT) kann aus dem Reflexionsspektren das Absorptionsspektrum der Probe berechnet werden, was eine bessere Auswertung der Probe ermöglicht.Dies ist aber häufig nur eingeschränkt möglich.
Neben dem oben beschriebenen Verfahren existiert noch eine weitere Reflexionsmethode, die vor allem für dünne Schichten und Adsorbaten geeignet ist, die sogenannte Technik der „Reflexion und Absorption“ (engl.: infrared reflection absorption spectroscopy, IRRAS). Hierbei wird IR-Strahlung an einer Metall-Oberfläche (extern) reflektiert. Durch die Metalloberfläche wird eine sehr hohe Reflektivität erreicht, zudem kommt es ähnlich wie bei der ob Oberflächenplasmonenresonanzspektroskopie zu einer Feldverstärkung, dies math die Methode selbst Monolagen, also dünne Schichten mit einer Schichtdicke von einigen Ångström, sensitiv (das können jedoch auch andere IR-Techniken). Die Messung erfolgt in der Regel in quasistreifenden Einfall, das heiß mit einem Einfallswinkel von rund 80° vom Lot. Des Weiteren kann durch den Einsatz von p-polarisiertem Licht kann die Sensitivität der Methode kann gesteigert werden.
Diffuse Reflexion
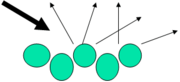 Prinzip der diffusen Reflexionsmessung
Prinzip der diffusen Reflexionsmessung- → Hauptartikel: DRIFTS
Mit der diffuse Reflexionsspektroskopie (DRIFTS) können rauhe Festkörper und Pulver untersucht werden, ohne letztere in eine Matrix zu pressen (vgl. Transmissionmessungen). Der Infrarotstrahl wird auf die Probe geführt und dort aufgrund der rauen Oberfläche nicht mehr gerichtet (wie bei einem Spiegel) sondern diffus reflektiert. Die reflektierte Strahlung wird von einer speziellen Spiegelanordnung, halbkugelförmig über der Probe (vgl. Hohlspiegel), wieder gebündelt und zum Detektor geführt.
Durch die diffuse Reflexion sind die Strahlenanteile ungleich verteilt und das entstehende Spektrum weißt Besonderheiten auf, die einen direkten Vergleich mit Spektren anderer IR-Techniken nur sehr eingeschränkt zulässt. Für eine Auswertung (qualitativ und quantitativ) kann das Spektrum mithilfe der Kubelka-Munk-Funktion in ein Absorptionsspektrum umgewandelt werden.
Abgeschwächte Totalreflexion (ATR)
 Halterung für ATR-Spektroskopie mir KRS5-Kristall
Halterung für ATR-Spektroskopie mir KRS5-Kristall- → Hauptartikel: ATR-Spektroskopie
Bei ATR-Spektroskopie wird die Strahlung in einem Reflexionselement (quasi ein Lichtwellenleiter) in Totalreflexion geführt. Dabei bildet sich an der Grenzfläche des Elements ein evaneszentes Feld aus, dessen Amplitude senkrecht zur Grenzfläche exponentiell abklingt (evaneszente Welle). Befindet sich hinter der Grenzfläche eine Probe wechselwirkt das evaneszente Feld mit der Probe, das heißt, bestimmte Frequenzbereiche können durch die Probe absorbiert werden. Die absorbierten Bereiche fehlen nun im Spektrum des reflektierten Strahls, der zum Detektor geführt wird. Da das Feld recht schnell abklingt, müssen Proben für die Messung sehr nah an die Oberfläche gebracht werden, um noch auswertbare Bandenintensitäten zu erreichen (der gemessene Bereich umfasst meist nur die ersten Mikrometern der Probe). Es ergeben sich ähnliche Spektren wie bei der Transmissionsspektroskopie, die jedoch im Vergleich dazu wellenlängenabhängige Intensitätsunterschiede aufweisen. Diese Methode eignet sich für feste und flüssige Proben.
Emission
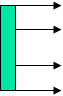 Prinzip der Emissionsmessung
Prinzip der EmissionsmessungBei der Infrarotemissionsspektroskopie (engl. infrared emission spectroscopy, IRES) wird eine Probe (meist über 100 °C) erhitzt und die emittierte Strahlung gemessen (vgl. Schwarzer Körper). [5][6]
Siehe auch
Einzelnachweise
- ↑ BGR Infrarot-Spektroskopie. Abgerufen am 17. Jul. 2008.
- ↑ C. Christiansen: Untersuchungen über die optischen Eigenschaften von fein verteilten Körpern - Erste Mittheilung. In: Annalen der Physik und Chemie. 23, 1884, S. 298–306.
- ↑ C. Christiansen: Untersuchungen über die optischen Eigenschaften von fein verteilten Körpern - Zweite Mittheilung. In: Annalen der Physik und Chemie. 24, 1885, S. 439–446.
- ↑ C. V. Raman: The theory of the Christiansen experiment. In: Proc. Ind. Acad. Sci.. 29, 1949, S. 381–390.
- ↑ S. Zhang, F. S. Franke, T. M. Niemczyk: Emission Spectroscopy. In: Francis Mirabella, Mirabella (Hrsg.): Modern Techniques in Applied Molecular Spectroscopy. 1. Auflage. Wiley & Sons, 1998, ISBN 0-471-12359-5, S. 323–376 (Einführung mit Beschreibungen zu verschiedenen Techniken).
- ↑ W. Suëtaka: Surface Infrared and Raman Spectroscopy: Methods and Applications. 1 Auflage. Springer-Verlag, New York 1995, ISBN 0306449633, S. 163ff. (Google Books).
Literatur
- Helmut Günzler, Hans-Ulrich Gremlich: IR-Spektroskopie: Eine Einführung. 4. Auflage. Wiley-VCH, Weinheim 2003, ISBN 978-3527-30801-9.
- M. Hesse, H. Meier, B. Zeeh: Spektroskopische Methoden in der organischen Chemie. 6. Auflage. Thieme, Stuttgart 2002, ISBN 3-13-576107-X.
- Bernhard Schrader: Infrared And Raman Spectroscopy. VCH, 1995, ISBN 3-527-26446-9.
Weblinks







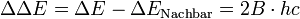




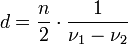



Wikimedia Foundation.