- MTP-AES
-
Atomspektroskopie (auch Atomspektrometrie) ist ein Sammelbegriff für spektroskopische Verfahren, die zur quantitativen und qualitativen Bestimmung von Elementen eingesetzt werden. Die Atomspektroskopie ist ein Teilbereich der Analytischen Chemie. Man unterscheidet
- Atomabsorptionsspektrometrie (AAS, High-Resolution Continuum Source AAS)
- Atomemissionsspektrometrie mit Flammen (F-AES)
- Optische ICP-Emissionsspektrometrie (ICP-OES)
- ICP-Massenspektrometrie (ICP-MS)
- Optische Emissionsspektrometrie mit Funken und Bogen (hier nicht behandelt)
- Röntgenfluoreszenzspektrometrie (hier nicht behandelt)
- Atomfluoreszenzspektroskopie (AFS)
Inhaltsverzeichnis
AAS (Atomabsorption)
 AAS-Spektrometer
AAS-SpektrometerDie Atomabsorptionsspektrometrie (AAS) ist eine bewährte und schnelle Methode zur quantitativen Analyse und qualitativen Analyse vieler Elemente (Metalle, Halbmetalle) in (meist) wässrigen Lösungen und Feststoffen.
Man unterscheidet in:
- F-AAS (Flammen-Atomabsorptionsspektrometrie, auch Flammen-Technik genannt)
- GF-AAS oder etA-AAS (Graphite Furnace-AAS; Atomabsorptionsspektrometrie mit elektrothermischer Aufheizung auch Graphitrohr-Technik genannt)
- CV-AAS (Cold Vapour-AAS; auch Hydrid-Technik genannt)
- HR-CS AAS (Atomabsorptionsspektrometrie mit Kontinuumstrahler und hochauflösendem Echelle-Doppelmonochromator)
Prinzip
 Kalibrierung in der AAS am Beispiel FIAS-Furnace-Technik
Kalibrierung in der AAS am Beispiel FIAS-Furnace-TechnikEine Lichtquelle emittiert Licht verschiedener Wellenlängen mit einer bestimmten Intensität. Im Strahlengang befindet sich eine Atomisierungseinheit, in der die Bestandteile einer zu untersuchenden Probe atomisiert, d. h. in einzelne, anregbare Atome überführt werden. Die Atomisierung der Elemente erfolgt entweder durch eine Gasflamme (Ethin/Luft- oder Ethin/Lachgas-Gemisch), in die die zu analysierende Lösung zerstäubt wird oder durch schnelles, starkes Erhitzen in einem elektrisch beheizten Graphitrohr, in das zuvor eine geringe Menge der Lösung hineingegeben wurde.
Nach Schwächung des Lichtstrahls in der Atomwolke (Absorption) wird seine Intensität hinter der Atomisierungseinheit gemessen und mit der Intensität des ungeschwächten Lichtes verglichen. Es wird detektiert, wie viel des eingestrahlten Lichtes einer bestimmten Wellenlänge durch das zu messende Element absorbiert wurde (in den meisten Fällen ist die AAS eine Einelementtechnik). Es gilt das Lambert-Beer'sche Gesetz. Mit steigender Konzentration des Analyten in der Probe steigt die Schwächung des eingestrahlten Lichtes (Extinktion) proportional.
Die absorbierte Licht-Energie wird vom dadurch angeregten Atom auf der gleichen Wellenlänge wieder abgestrahlt: Atom-Fluoreszenz. Dass man eine Intensitäts-Schwächung, das Absorptions-Signal, messen kann, hat einen geometrischen Grund: Das eingestrahlte Licht wird durch die Optik des Gerätes auf einen sehr kleinen Raumwinkel fokussiert. Die Re-Emission erfolgt jedoch als Kugelwelle über den gesamten Raumwinkel von 4π. Nur ein vernachlässigbar kleiner Anteil davon gelangt mit dem Licht der Lampe durch den Austritts-Spalt.
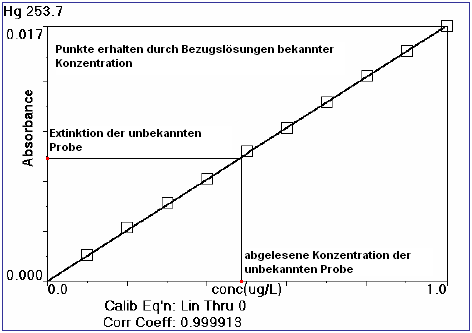
Die AAS ist ein relatives Messverfahren. Nach dem Lambert-Beer'schen Gesetz (gültig für niedrige Konzentrationen) wird die Extinktion von Kalibrierungs-Standards bekannter Konzentrationen aufgenommen, eine Kalibrierkurve erstellt und Proben mit unbekannter Konzentration gegen diese Kalibrierung aufgenommen und die Konzentration abgelesen (heutzutage per Computersoftware ausgewertet). Ein großer Vorteil der AAS gegenüber anderen spektroskopischen Verfahren ist seine Selektivität. Die als Lichtquellen eingesetzten Lampen emittieren aufgrund der Zusammensetzung ihres Leuchtmittels (Hohlkathodenmaterial, Salz in einer elektrodenlosen Entladungslampe (EDL)) ein elementspezifisches elektromagnetisches Spektrum, das gezielt von dem gleichen, zu untersuchenden Element absorbiert wird. Spektrale Störungen kommen in der AAS nur sehr selten vor. Neueste auf dem Markt erhältliche Entwicklungen wie High-Resolution Continuum Source AA-Spektrometer arbeiten hingegen mit nur einer Lichtquelle. Eine Xenon-Kurzbogenlampe als kontinuierliche Strahlungsquelle deckt alle Elemente und alle verfügbaren Wellenlängen ab. Diese Strahlungsquelle eröffnet den gesamten für die AAS relevanten Wellenlängenbereich in nur einem Schritt. Damit ist die sequentielle Multielementroutine möglich. Ein Novum sind für die Auswertung nutzbare Molekülbanden, mit denen zusätzliche Elemente, wie z. B. Schwefel oder auch Phosphor, analysiert werden können. Ein HR-CS AAS misst daher unabhängig von Hohlkatodenlampen. Das bietet Vorteile wie z. B. geringere Vorbereitungszeiten, keine langwierige Einbrennzeit der Lichtquelle, da Drifterscheinungen simultan korrigiert werden.
Aufbau
Linienstrahler

Lichtquelle → Atomisierungseinheit → Dispersionseinheit (Monochromator) → Detektor (Photomultiplier, Halbleiterdetektor)
Lichtquellen
In der AAS werden elementspezifische Lampen verwendet. Man unterscheidet zwischen
 Hohlkathodenlampe
Hohlkathodenlampe- Hohlkathodenlampen mit einer Kathode bestehend aus dem Element des Analyten. Alternativ können auch
- Superlampen (zusätzliche Kathode) oder
- Elektrodenlose Entladungslampen (EDL, Prinzip Gasentladungslampe) eingesetzt werden.
Beide zuletzt genannten Lampentypen bieten eine höhere Lichtintensität, was insbesondere Elementen, die im UV-Bereich absorbieren (As, Cd, Pb, Sb, Se, Bi, Tl, Hg), eine bessere Nachweisgrenze durch besseres Signal-zu-Rauschen-Verhältnis (S/N ratio) beschert. Normale Hohlkathodenlampen zeigen unterhalb etwa 300 nm eine deutliche Intensitätsverschlechterung ihrer Emissionslinien. Beide Lampentypen erfordern eine separate Spannungsversorgung, die bei modernen Geräten aber z. T. schon eingebaut ist. In der HR-CS AAS verwendet man nur eine einzige Strahlungsquelle, eine speziell entwickelte Xenon-Kurzbogenlampe als kontinuierliche Strahlungsquelle für alle Elemente und alle Wellenlängen.
Die Lampe hat eine veränderte Elektrodenform und arbeitet unter hohem Druck. Unter diesen Bedingungen bildet sich ein heißer Brennfleck aus, der eine Temperatur von etwa 10000 K erreicht. Die Emissionsintensität dieser Lampe ist über den gesamten Spektralbereich um mindestens Faktor 10, im fernen UV um mehr als Faktor 100 intensiver als die von konventionellen Xenon-Kurzbogenlampen. Und was für die AAS vielleicht noch wichtiger ist, die Emissionsintensität über den gesamten Spektralbereich ist im Durchschnitt um Faktor 100 intensiver als die von üblichen Hohlkathodenlampen. Einer der großen Vorteile der HR-CS AAS ist sicher, dass nur eine einzige Strahlungsquelle für alle Elemente und alle Wellenlängen über den gesamten Spektralbereich von 190 – 900 nm erforderlich ist. Ein weiterer Vorteil resultiert aus der weitaus höheren Emissionsintensität dieser Lampe. Die Strahlungsintensität hat in der AAS zwar keinen Einfluss auf die Empfindlichkeit, wohl aber auf das Signal/Rausch-Verhältnis.
Atomisierung
Das Ziel bei der Atomabsorptionsspektroskopie besteht darin, einen möglichst hohen Anteil von Atomen in den gasförmigen Aggregatzustand zu überführen und möglichst wenig angeregte oder ionisierte Atome zu erzeugen . Dazu muss die Probe verdampft (frei von Lösemitteln und leicht flüchtigen Bestandteilen) und verascht werden und in freie Atome dissoziieren. Zur Atomisierung werden in der AAS vorwiegend Flammen und Graphitrohröfen eingesetzt. Dabei muss zwischen F-AAS und GF-AAS unterschieden werden. Bei der F-AAS wird die Probe kontinuierlich mit einer konstanten Geschwindigkeit zugeführt, woraus man zeitlich konstante Signale erhält. Bei der GF-AAS wird nur einmal eine bekannte Probemenge aufgegeben. Im Idealfall hat das spektrale Signal ein Maximum und fällt dann auf Null ab, wenn die Atomwolke aus dem Atomisator herausgetragen wird. In der HR-CS AAS werden die gleichen Atomisatoren eingesetzt wie in der klassischen Linienstrahler-AAS. Wegen der Sichtbarkeit der spektralen Umgebung der Analysenlinie ist in der HR-CS AAS die Methodenentwicklung und -optimierung signifikant erleichtert und vereinfacht.
F-AAS (Flammen-AAS)
 "normale" Lachgasflamme
"normale" Lachgasflamme Lachgasflamme bei hoher Salzfracht
Lachgasflamme bei hoher SalzfrachtBei der Flammentechnik wird die gelöste Probe zunächst in ein Aerosol überführt. Dazu wird die Probe mit einem pneumatischen Zerstäuber in eine Mischkammer hinein zerstäubt und mit Brenngas und Oxidans verwirbelt. Es bildet sich ein feiner Nebel, ein Aerosol. Um die Tropfengröße noch kleiner und gleichmäßiger zu machen, trifft das Aerosol zunächst auf eine Prallkugel aus Keramik und anschließend ggf. auf einen Mischflügel, der nur feine Tröpfchen passieren lässt. Ein geringer Teil des ursprünglichen Aerosols gelangt schließlich aus der Mischkammer in die Flamme. Dort verdampft zunächst das Lösungsmittel und die festen Probenbestandteile schmelzen, verdampfen und dissoziieren schließlich. Zu hohe Flammentemperaturen können insbesondere bei Alkali- und einigen Erdalkalielementen zu Ionisationsinterferenzen führen, die durch Zugabe eines Ionisationspuffers (CsCl oder KCl) kontrolliert werden können. Zu niedrige Flammentemperaturen führen zu chemischen Interferenzen. In der Flammen-AAS kann die Flamme alternativ mit zwei unterschiedlichen Gasgemischen betrieben werden.
- Luft-Acetylen-Flamme: In der Regel wird diese Flamme eingesetzt, sie verwendet Luft als Oxidans und Acetylen als Brenngas.
- Lachgas-Acetylen-Flamme: Die Verbindungen einiger Elemente (z. B. Al, Si, Ti, aber auch Ca und Cr) erfordern höhere Temperaturen zur Dissoziation. In diesem Fall wird anstelle von Druckluft das Gas Distickstoffoxid (Lachgas) als Oxidans eingesetzt. Diese Flamme ist mit ca. 2800 °C um ca. 500 °C heißer als die Luft-Acetylen-Flamme. Durch ihre reduzierende Wirkung können auch Oxide von z. B. Cr, Ca und Al atomisiert werden.
GF-AAS (Graphitrohr-AAS) oder EtA-AAS (AAS mit elektrothermischer Aufheizung)
Bei der Graphitrohr-AAS macht man sich dem Umstand zu nutze, dass Graphit den Strom leitet und sich beim Anlegen einer elektrischen Spannung durch seinen elektrischen Widerstand erhitzt.
Zunächst werden 5 bis 50 Mikroliter der Probelösung in ein Graphitrohrofen gebracht und in mehreren Schritten erhitzt. Das Programm hängt wesentlich von dem zu analysierenden Element sowie seiner chemischen Umgebung ab. Außerdem spielt es eine große Rolle, in was für einem Gerät und in was für einem Graphitrohrofensystem (längsbeheizter/querbeheizter Graphitrohrofen) gearbeitet wird. Generell kann gesagt werden, dass im querbeheizten Graphitrohrofen ca. 200 °C geringere Pyrolysetemperaturen und 200 bis 400 °C geringere Atomisierungstemperaturen eingesetzt werden. Als Anhaltspunkt für die Wahl des richtigen Temperatur-/Zeitprogramms sollten die "Empfohlenen Bedingungen" des Graphitrohrofenherstellers dienen. Hiervon ausgehend sollten Temperaturen und Zeiten so optimiert werden, dass das Messsignal bei minimalem Untergrundsignal eine maximale Signalfläche erhält. Die Probenzusammensetzung kann eine Abweichung von Standardprogramm erforderlich machen.
- Trocknung 1: für etwa 30 s wird der Ofen auf 90 bis 130 °C geheizt um die Probe einzuengen und nahezu zu trocknen.
- Trocknung 2: für etwa 20 s wird der Ofen auf 400 °C geheizt um die Probe vollständig zu trocknen (wenn Kristallwasser vorhanden)
- Pyrolyse: für etwa 30 s wird der Ofen auf 400 °C bis 1500 °C (abhängig vom Element) geheizt um die organische Bestandteile zu entfernen. Dies geschieht durch Pyrolyse oder Veraschung
- Atomisierung: bei 1500 bis 2500 °C (abhängig von der elementspezifischen Atomisierungstemperatur) wird die Probe etwa 5 s atomisiert
- Ausheizen: schließlich wird nach Ende der Analyse noch etwa 3 s auf 2500 °C (querbeheizter Ofen) bis 2800 °C (längsbeheizter Ofen) geheizt, um Restbestände der Probe zu atomisieren
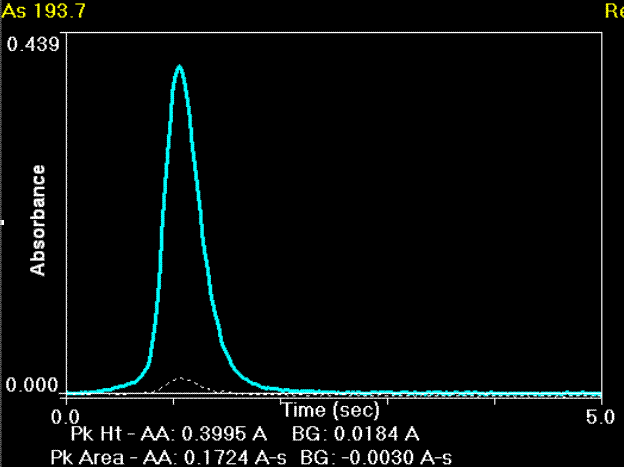 transientes (zeitaufgelöstes) Signal einer Messung von Arsen im Graphitrohrofen
transientes (zeitaufgelöstes) Signal einer Messung von Arsen im Graphitrohrofen glühende Plattform im längsbeheizten Graphitrohrofen
glühende Plattform im längsbeheizten Graphitrohrofen querbeheizter Zeeman-Graphitrohrofen
querbeheizter Zeeman-Graphitrohrofen
Jeder Schritt beinhaltet eine Anstiegszeit (Rampe), innerhalb derer die angegebene Temperatur erreicht wird. Je langsamer die Aufheizrate gewählt wird, desto geringer ist die Gefahr eines Verspritzens von Probe und desto besser wird die Präzision bei mehreren Wiederholmessungen.Für eine schonendere Trocknung kann der Schritt "Trocknung 1" aufgeteilt werden, z. B. in einen Schritt bei 110°C und einen Schritt bei 130°C. Schritt "Trocknung 2" kann bei einfachen Proben (Trinkwasser) auch wegfallen; er wird eher bei Proben mit komplexer Matrix (Körperflüssigkeiten oder stark salzhaltige Abwässer) eingesetzt. Für den Atomisierungsschritt wählt man üblicherweise eine Rampe von 0 Sekunden, hierbei wird die maximale Leistung der Spannungsversorgung auf das Graphitrohr gegeben, um eine maximale Aufheizrate zu erzielen. Dadurch erreicht die Atomwolke des Analyten eine maximale Dichte und es ergibt sich eine maximale Empfindlichkeit. Die Temperaturen sind natürlich abhängig vom Analyten und können stark abweichen. Vorteilhaft gegenüber der Flammtechnik ist, dass die Probe quantitativ in den Strahlengang gebracht werden kann und dort auch länger (bis zu 7 s) verbleibt. Weiter können oft störende Matrixbestandteile durch unterschiedliche Verdampfungstemperaturen abgetrennt werden; entweder verdampfen sie vorher, oder sie bleiben zurück. Die Nachweisgrenzen sind daher bis zu 3 Zehnerpotenzen besser als bei der Flammentechnik oder der ICP-OES. Allerdings kann es zu Interferenzen kommen, wenn nicht unter spezifischen Messbedingungen gearbeitet wird. Die Zusammenfassung aller Maßnahmen, die zu einer störungsfreien Analytik in der Graphitrohr-AAS führen, wird als STPF-Konzept (Stabilized Temperature Platform Furnace) bezeichnet.
STPF-Konzept
- Pyrolytisch beschichtetes Graphitrohr (bessere Haltbarkeit und Empfindlichkeit)
- Plattform im Graphitrohr (Atomisierung in eine temperaturkonstante Gasphase im Graphitrohr)
- Peakflächenauswertung (geringere Abhängigkeit vom Zeitpunkt der maximalen Atomisierung des Analyten)
- Einsatz von Modifiern (Stabilisierung des Analyten bei höheren Pyrolysetemperaturen oder Verringerung der Zersetzungstemperatur von Matrixbestandteilen)
- Gas-Stop während der Atomisierung (Atomwolke verbleibt länger im Rohr)
- schnelle Signalerfassung
- querbeheizter Graphitrohrofen (Temperaturkonstanz über die gesamte länge des Rohres, keine Kondensationseffekte und Rekombination zu Molekülen)
Hydrid- und Kaltdampftechnik
Begriffsdefinition
- HG-AAS Hydride Generation-AAS Hydridtechnik
- CV-AAS Cold Vapour-AAS Kaltdampftechnik
- HG-ET-AAS Hydride Generation Electrothermal-AAS Anreicherung von Hydriden im Graphitrohrofen (auch: FIAS-Furnace, FIFU)
Hydridtechnik
 elektrisch beheizte Quarzzelle bei 1000 °C zur Bestimmung von Arsen
elektrisch beheizte Quarzzelle bei 1000 °C zur Bestimmung von ArsenBei einigen Elementen, vornehmlich Zinn, Arsen, Antimon, Bismut, Selen, Tellur und Germanium können mit der relativ einfachen Hydridtechnik vergleichbare Nachweisgrenzen wie im Graphitrohrofen erreicht werden. Wenn das zu bestimmende Element mit naszierendem Wasserstoff gasförmige Hydride wie zum Beispiel AsH3, SnH4 oder H2Se bildet, können diese durch Inertgas (i. d. R. Argon) aus ihrer Lösung getragen und in eine beheizte Glasküvette überführt werden. Die Küvette besteht aus Quarzglas, da einfaches Glas bei der verwendeten Temperatur (bis 1000 °C) mit der Zeit ausglast. Die Heizung kann entweder eine Elektroheizung oder die Flamme eines Flammen-AAS sein. Ein Vorteil der Elektroheizung besteht in der besseren Temperaturregulierung, da nicht jedes Element sein Empfindlichkeitsmaximum bei der gleichen Temperatur hat. In der Zelle zerfallen die Hydride bei Temperaturen um die 1200 K wieder in Wasserstoff und das zu bestimmende Element. Diese Reaktion ist nicht nur temperaturgesteuert, sondern sie hängt auch von der Oberflächebeschaffenheit der Zelle ab. Die Hydridtechnik beschränkt sich nicht ausschließlich auf die AAS, sie findet auch in der ICP-OES ihre Anwendung.
Kaltdampftechnik
Als Unterform der Hydridtechnik ist die Kaltdampftechnik (CV-AAS) aufzufassen. Hierbei wird mit Hilfe eines Reduktionsmittels kein Hydrid, sondern atomares Quecksilber generiert. Das Reduktionsmittel kann, wie oben, Natriumborhydrid NaBH4 sein, häufiger wird jedoch Zinn (II) chlorid verwendet, das eine höhere Empfindlichkeit bietet und weniger zu Schaumbildung neigt. Bei Quecksilber spricht man von Kaltdampftechnik, da die Quarzzelle nicht beheizt sein muss und keine Aktivierungsenergie für eine Zersetzung des Hydrids benötigt wird. Trotzdem ist ein leichtes Erwärmen auf 50 bis 100 °C vorteilhaft, damit sich kein Wasserdampf in der Küvette absetzt, was die Empfindlichleit stören kann.
FIAS-Technik
Wenn Hydrid bzw. Quecksilber durch ein Fließinjektionssystem erzeugt wird, können viele Proben schnell hintereinander mit wenig Verschleppung zwischen den Proben analysiert werden. Hierbei wird ein definiertes Probenaliquot über ein Ventil in einen Reaktor gepumpt, mit Reduktionslösung versetzt und mit Argon durchmischt. Vor und nach dem Probenaliquot wird das System durch Umschalten des Ventils mit Säure gespült. In einem Gas-Flüssigseparator entweicht das entstandene Gas (Hydrid oder Quecksilberdampf), verbliebene Flüssigkeit wird über einen Abfallschlauch abgepumpt. Das Gas wird in die Quarzzelle überführt und mit dem Spektrometer gemessen.
FIAS-Furnace-Technik
Die Zersetzung der Hydride kann jedoch nicht nur in der Quarzzelle, sondern auch im Graphitrohr erfolgen (Hydride Generation Electrothermal-AAS, HG-ET-AAS). Hierfür muss das Rohr einmalig mit einer Schicht aus Iridium (Lösung von IrCl3) belegt werden, an dessen Oberfläche sich das Hydrid zersetzt und der Analyt (auch Quecksilber) anreichert. Über ein kurzes Temperaturprogramm wird der Analyt atomisiert, ohne die Beschichtung zu zerstören. Die Nachweisstärke liegt, bei gleichem Probenvolumen, im Bereich der herkömmlichen Hydridtechnik, kann jedoch durch größere Volumina beliebig gesteigert werden. Vorteil ist die geringere Störanfälligkeit bei der Zersetzung der Hydride, die in der Quarzzelle stark von der Oberflächenbeschaffenheit der Zelle (Stoßreaktion) und weniger von der Temperatur abhängt (solange die Aktivierungsenergie ausreicht). Die Technik wird auch, in Kombination mit einem Fließinjektionssystem, als FIAS-Furnace-Technik, FIFU, bezeichnet.
Interferenzen in der AAS
Durch die Anwesenheit von Begleitsubstanzen in der Probe kann es zu Störungen (Interferenzen) kommen. Man unterscheidet:
- spektrale Interferenzen
- nicht spektrale Interferenzen
Spektrale Interferenzen werden bis zu einem bestimmten Grad durch Untergrundkorrektur bereinigt oder zumindest verringert. Dazu wird in der AAS neben der Strahlungsquelle zusätzlich eine Deuterium- (D2-) Lampe in den Strahlengang geschaltet oder alternativ in der Graphitrohrofen-AAS die Zeeman-Untergrundkorrektur verwendet. Die High-Resolution Continuum Source AAS arbeitet mit einem hochauflösendem Echelle-Spektrometer. Neben der Intensität der Analysenlinie wird auch die spektrale Umgebung simultan registriert. Dadurch sind Interferenzen sofort sichtbar. Die Notwendigkeit einer Optimierung oder Korrektur der Parameter werden in den HR-CS AA-Spektrometern automatisch erkannt. Auch durch den Einsatz eines leistungsstarken Detektors wird bei optimaler Linientrennung eine Minimierung der Interferenzen erreicht.
Deuterium (D2)-Untergrundkorrektur
Bei Verwendung einer Deuteriumlampe für die Korrektur des Untergrundes wird die Lichtschwächung der Hohlkathodenlampe und die einer D2-Lampe entweder zeitgleich oder abwechselnd erfasst. Die D2-Lampe liefert, im Gegensatz zur Hohlkathodenlampe, ein kontinuierliches Lichtspektrum, dessen Intensität von der Wellenlänge abhängt. Oberhalb von ca. 350nm liefert sie so gut wie keine Intensität, so dass Elemente mit einer Absorptionslinie oberhalb dieser Wellenlänge ohne Untergrundkorrektur gemessen werden können. Die Auswahl der Wellenlänge (besser des Wellenlängenbereiches) zur Bestimmung der Untergrundabsorption erfolgt über die Breite des Monochromatorspaltes am Spektrometer. In erster Näherung wird das Licht der Deuteriumlampe fast nur durch den Untergrund absorbiert. Der Anteil der Schwächung der Analysenwellenlänge ist im Vergleich zur Schwächung der übrigen, vom Spalt durchgelassenen Wellenlängen, vernachlässigbar gering.
Bei der Auswertung wird von der gemessenen Strahlung der Hohlkathodenlampe (Gesamtabsorption aus Untergrund + Atomabsorption) die Absorption der Strahlung der D2-Lampe (näherungsweise nur Untergrundabsorption) abgezogen. Man erhält die Absorption des Analyten in der Probe. Ein prinzipieller Fehler liegt in der Messung des Untergrundes innerhalb eines Wellenlängenbereiches, vorgegeben durch die Einstellung des Spaltes im Monochromator, und nicht exakt auf der Analysenwellenlänge. Wenn also der Untergrund besonders stark neben der eigentlichen Analysenlinie absorbiert, wird ein zu großer Betrag Untergrundabsorption von der Gesamtabsorption abgezogen. Es kommt zu einer sog. "Überkorrektur" des Messsignals mit negativen Messergebnissen.
Zeeman-Untergrundkorrektur
Das Magnetfeld des Zeeman-Magneten kann man als zweite "Strahlungsquelle" auffassen. Bei ausgeschaltetem Magnetfeld wird die gesamte Lichtschwächung von Analyt + Untergrund aufgenommen. Bei eingeschaltetem Magnetfeld erfolgt die Zeeman-Aufspaltung der Absorptionslinie, so dass jetzt der Analyt nicht mehr auf der von der Lampe emittierten Wellenlänge absorbiert, sondern nur noch die Matrix (der Untergrund). Die Stärke des angelegten Magnetfeldes reicht nicht für eine Zeeman-Aufspaltung von Molekülen oder Teilchen (dem Untergrund) aus. Der Vorteil dieser Untergrundkorrektur liegt in der Messung des Untergrundes exakt auf der Analysenlinie, wodurch das Untergrundsignal kleiner ist und auch bei höherer Salzfracht noch störungsarm gemessen werden kann. Nachteil ist ein verringerter Linearitätsbereich und, je nach Element, eine verringerte Empfindlichkeit durch z. T. unvollständige Zeeman-Aufspaltung.
Ursachen für spektrale Interferenzen in der AAS sind:
- Emission der Flamme oder des glühenden Graphitrohrofens (sog. Gleichlicht),
- ungewünschte Absorption auf der gleichen Wellenlänge
- Streuung an festen, schwer verdampfbaren Rauchpartikeln und Gasen (Rayleigh-Streuung)
Untergrundkorrektur in der HR-CS AAS
In der HR-CS AAS wird kein zusätzliches System zur Untergrundkorrektur benötigt. Diese Geräte sind mit einer CCD-Zeile und damit im Prinzip simultan und unabhängig arbeitenden Detektoren ausgestattet. Von der Software werden einige dieser Detektoren auf beiden Seiten der Analysenlinie ausgewählt und für Korrekturzwecke eingesetzt. Jede Änderung in der Strahlungsintensität, die auf allen Korrekturpixel gleichermaßen auftreten, werden automatisch korrigiert. Hierzu gehören z. B. Schwankungen in der Lampenemission, aber auch jegliche kontinuierliche Untergrundabsorption. Diskontinuierliche Untergrundabsorption, z. B. direkte Linienüberlagerung mit einem Matrixelement oder Molekülabsorption mit Feinstruktur, kann mit Hilfe von Referenzspektren rechnerisch beseitigt werden. Es können breitbandige und spektrale Untergrundeffekte getrennt werden. Erstere werden automatisch über Referenzpixel korrigiert und zweitere werden sichtbar und damit bewertbar gemacht. In den meisten Fällen von spektralen Interferenzen ist die hervorragende Auflösung schon ausreichend, so dass die Analysenlinie ungestört zur Auswertung herangezogen werden kann. Mit dieser Technik wird der Arbeitsablauf gerade bei unbekannten und wechselnden Proben extrem vereinfacht. Aber auch bei Routinemessungen mit bekannter Matrix wird die Messroutine erleichtert, da spektrale Störungen nicht mehr aufwendig korrigiert werden müssen. Vollautomatisch ablaufende Untergrundroutinen nutzen die zur Verfügung stehenden Referenzpixel und ermöglichen eine zeitechte simultane Korrektur. Die Untergrundkorrektur in der HR-CS AAS bietet einen großen dynamischen linearen Arbeitsbereich, erweiterte Nachweisgrenzen, eindeutige Messergebnisse, eliminiert Artefakte und korrigiert bei direkter Linienüberlagerung.Nicht spektrale Interferenzen
Nicht spektrale Interferenzen entstehen beim Atomisierungsvorgang. Man unterscheidet:
- Transportinterferenzen sind chemische Störungen durch Matrixkomponenten oder durch physikalische Störungen durch die Viskosität, Dichte oder der Oberflächenspannung des Lösungsmittels. Sie sind besonders in der Flammen–AAS problematisch, da hier nur ein sehr geringer Anteil der Probe in die Flamme gelangt. Die Eliminierung von Transportinterferenzen wird durch das Standard-Additionsverfahren oder eine Matrixanpassung der Kalibrierungs-Standards an die Probe erreicht.
- Gasinterferenzen entstehen, wenn es nicht zur vollständigen Dissoziation (AB-> A+B) oder zu einer Ionisierung kommt. Durch Zugabe von Freisetzungsmitteln, beispielsweise LaCl3 für Phosphate, kann eine vollständige Dissoziation erreicht werden. Durch Zugabe von Alkalielementen kann eine bessere Ionisierung erreicht werden. In beiden Fällen gehorcht die Wirkungsweise dem Massenwirkungsgesetz. Ein Überschuss dieses Hilfsmittels bewirkt eine Verschiebung des Reaktionsgleichgewichtes zum gasförmigem Analyten im atomaren elektronischen Grundzustand.
- Verdampfungsinterferenzen spielen im Graphitrohr eine Rolle. Sie entstehen durch zu frühe oder zu späte Verdampfung des Analyten in der Probe, bezogen auf das Verhalten des Analyten im Kalibrierungs-Standard. Dadurch kann das Signal einer Probe und eines Kalibrierungs-Standards bei gleichem Analytgehalt eine deutlich unterschiedliche Signalform und -höhe ergeben. Ein zeitlich aufgelöstes Signal (Auswertung in Signalfläche) kann hier noch zu richtigen Ergebnissen führen, ohne auf eine Standardaddition zurückgreifen zu müssen.
Matrixmodifizierer in der GF-AAS
Hilfreich kann in diesem Zusammenhang auch die Verwendung von Matrixmodifizierern sein, die den Analyten in eine einheitliche, thermisch stabilere, chemische Verbindung überführen (Isoformierungshilfe). Dadurch sind während der Pyrolyse höhere Temperaturen möglich, um Matrix vor der Atomisierung zu entfernen, ohne den Analyten vorzeitig zu verlieren. Für die Bestimmung von Blei und Cadmium wird häufig ein Mischmodifizierer aus Mg (NO3)2 und NH4H2PO4 eingesetzt. Zur Bestimmung vieler anderer Elemente hat sich der Einsatz eines Mischmodifizierers aus Pd (NO3)2 und Mg (NO3)2 bewährt, der Pyrolysetemperaturen von etwa 1000 °C zulässt, das ist in querbeheizten Graphitrohrsystemen die Temperatur, bei der NaCl verflüchtigt wird, ein häufiger Bestandteil von Körperflüssigkeiten, Abwässern und Produkten der chemischen Industrie.
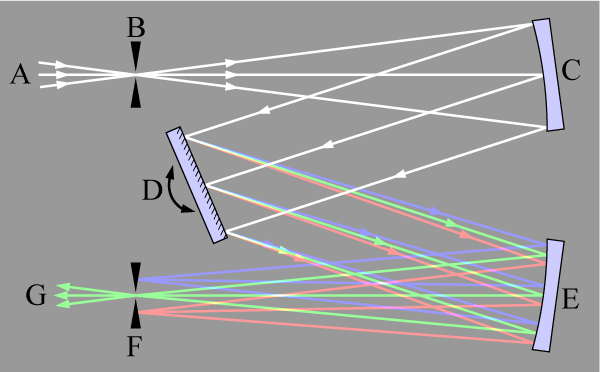
Monochromator
Der Monochromator teilt das aus Lampe und leuchtender Atomisierungseinheit stammende Licht in sein Spektrum auf und isoliert daraus eine bestimmte Wellenlänge. Bei modernen Geräten werden dazu ausschließlich holographische Gitter verwendet, die das einfallende Licht beugen. Je höher die Zahl der Furchen auf dem verwendeten Gitter ist, desto besser ist das Auflösungsvermögen der Optik eines Spektrometers. Im Gegensatz zu den Emissionstechniken werden in der AAS Linienstrahler als Strahlungsquellen eingesetzt, die kein kontinuierliches Spektrum erzeugen, sondern nur die Spektrallinien des darin enthaltenen Elementes. Aus diesem Grunde sind die Ansprüche an das Auflösungsvermögen der Optik eines AAS geringer als an die Optik eines ICP-OES. Über die Wahl der Breite des Austrittsspaltes wird der Wellenlängenbereich eingegrenzt, der auf den Detektor gelangt. Der Einsatz eines Kontinuumstrahlers in High-Resolution Continuum Source AA-Spektrometern erfordert zwangsläufig die Verwendung eines hochauflösenden Monochromators. Klassische Monochromatoren dieser Art, wie sie in der optischen Emission eingesetzt wurden, haben einen großen Platzbedarf und neigen stark zu Wellenlängendrift. Beides ist in der HR-CS AAS nicht akzeptabel. Das Problem wurde mit der Konstruktion eines kompakten Doppelmonochromators mit aktiver Wellenlängenstabilisierung gelöst. Beide Monochromatoren sind in Littrow-Aufstellung mit einer Brennweite von 30 bzw. 40 cm. Die Strahlung des Kontinuumstrahlers gelangt durch den Eintrittsspalt in den Monochromator und wird von dem ersten Parabolspiegel auf das Prisma umgelenkt. Das Prisma ist auf der Rückseite verspiegelt, so dass die Strahlung das Prisma zweimal passiert bevor sie, nun spektral zerlegt, wieder auf den Parabolspiegel fällt. Dieser führt die Strahlung über einen Umlenkspiegel zum Zwischenspalt. Das Prisma wird dabei so gedreht, dass die Strahlung im Bereich der Analysenlinie durch den Zwischenspalt in den zweiten Monochromator gelangt. Der zweite Parabolspiegel lenkt die Strahlung auf das Echellegitter, wo der ausgewählte Spektralbereich nun hoch aufgelöst wird. Das gesamte hochaufgelöste Teilstück des Spektrums wird dann von dem Parabolspiegel auf dem Detektor abgebildet. Die Auflösung des Doppelmonochromators liegt traditionell bei einem Wert, der etwa um einen Faktor 100 besser ist als die Auflösung klassischer AAS-Geräte.
Grundanordnungen von Monochromatoren- Czerny-Turner-Anordnung
- Echelle-Anordnung
- Littrow-Anordnung
Czerny-Turner
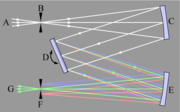 Czerny-Turner-Anordnung eines Monochromators
Czerny-Turner-Anordnung eines MonochromatorsLicht (A) wird auf den Eintrittsspalt fokussiert (B) und wird mit einem Konkavspiegel kollimiert (parallelisiert) (C). Der kollimierte Strahl wird durch ein drehbares Gitter gebeugt (D) und der dispergierte Strahl wieder durch einen zweiten Spiegel (E) auf den Austrittsspalt (F) fokussiert. Jede Wellenlänge des Lichtes wird auf eine andere Stelle des Spaltes fokussiert. Die Wellenlänge, die durch den Spalt (G) hindurchgelassen wird, hängt vom Drehwinkel des Gitters (D) ab.
Echelle
Echelle-Monochromatoren kombinieren in der Atomspektrometrie die Vorteile einer sehr kompakten Bauweise mit hohem Lichtdurchsatz bei sehr guter optischer Auflösung. Durch die Kombination eines Echellegitters (Aufspaltung des Lichtes in seine Beugungsordnungen) mit einem Prisma (Aufspaltung der Beugungsordnungen in die Wellenlängen) ergibt sich eine zweidimensionale Anordnung des Spektrums, die bei geeigneter Wahl des Detektors (Halbleiterdetektor mit Segmentierung der lichtempfindlichen Bereiche) eine simultane Erfassung mehrerer Wellenlängen zulässt. Realisiert wird diese Anordnung in vielen OES und in wenigen AAS.
Detektor
Zur Messung der Lichtschwächung setzt man Sekundärelektronenvervielfacher (SEV) – oder heutzutage vermehrt – Halbleiterdetektoren Solid State Detector (SSD) ein. Letztere zeigen eine homogenere und effektivere Lichtausbeute (Quanteneffizienz) über den interessierenden Wellenlängenbereich (190–900 nm) und damit ein besseres Signal/Rausch-Verhältnis, was sich in besseren Nachweisgrenzen widerspiegelt.
Als Detektor wird in der High-Resolution Continuum Source AAS eine CCD-Zeile verwendet. Jedes Pixel wird dabei unabhängig ausgewertet, so dass das Gerät im Prinzip mit unabhängigen Detektoren arbeitet. Alle Pixel werden simultan belichtet und simultan ausgelesen. Während der Signalverarbeitung erfolgt bereits die nächste Belichtung, was eine sehr rasche Messfolge ermöglicht. Die Absorptionslinie wird im Wesentlichen von fünf Pixel erfasst, während die übrigen Pixel lediglich die statistischen Schwankungen der Grundlinie zeigen. Da meist nur wenige Pixel zur Messung der Atomabsorption eingesetzt werden, können die übrigen für Korrekturzwecke verwendet werden. Nachdem alle Pixel simultan belichtet und ausgelesen werden, können alle Intensitätsschwankungen, die nicht wellenlängenabhängig sind, wie etwa Schwankungen in der Lampenemission, mit Hilfe von Korrekturpixel ermittelt und eliminiert werden. Dadurch entsteht ein extrem stabiles und rauscharmes System, das zu einer deutlichen Verbesserung des Signal/ Rausch-Verhältnisses führt. Das gleiche Korrektursystem eliminiert automatisch auch jegliche kontinuierliche Untergrundabsorption. Der Detektor registriert nicht nur die Strahlung auf der Analysenlinie, sondern ihre gesamte Umgebung. Damit werden z. B. spektrale Interferenzen erkennbar und können leichter vermieden werden.F-AES (Flammen-Emission)
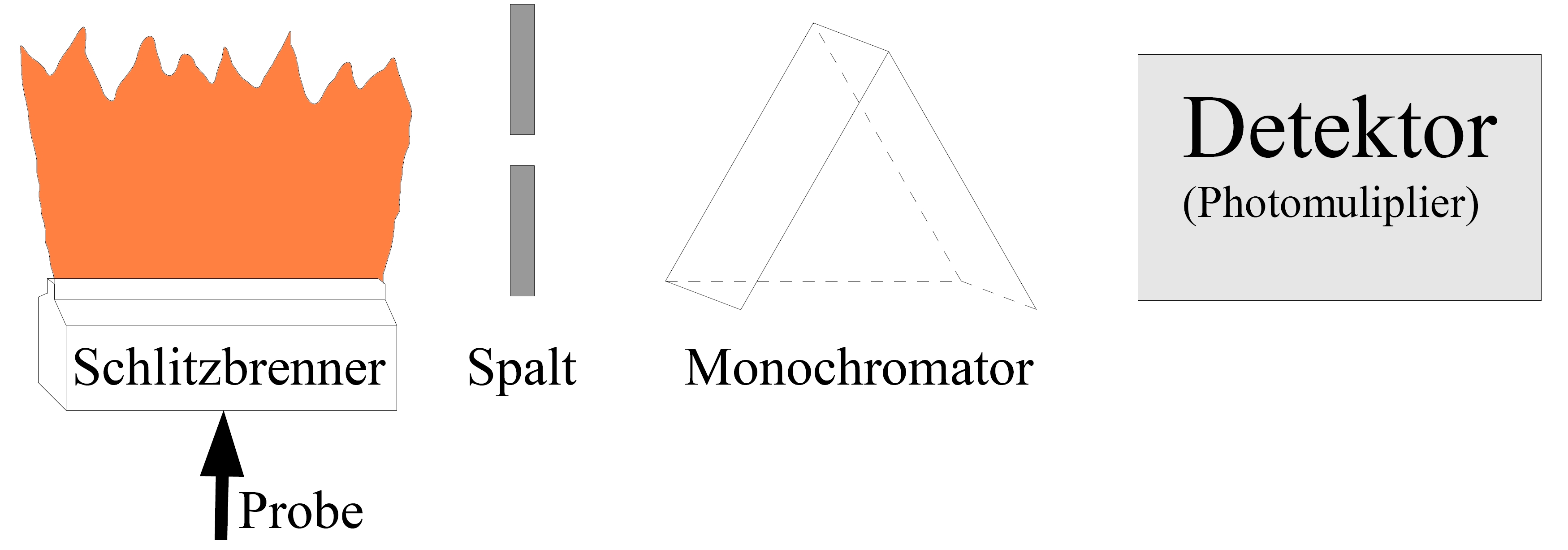
Flammen-Atomemissionsspektrometrie
Prinzip
Eine Materialprobe (Analysesubstanz) wird in die Flamme gebracht (z. B. als Lösung verdampft und der Dampf der Flamme zugeführt). Durch die Wärmeenergie der Flamme werden die äußeren Valenzelektronen angeregt und auf ein energetisch höheres Niveau gehoben. Beim Rückfall in den Grundzustand wird die vorher zugeführte Energie als Lichtenergie abgegeben; dabei emittieren die Atome ihr elementspezifisches Spektrum, das im Spektrometer dispergiert und analysiert wird. Das Flammenemissionsspektrum wird mit einem Flammenphotometer oder, da inzwischen häufiger am Markt vertreten, mit einem Flammen-Atomabsorptionsspektrometer im Emissionsbetrieb, gemessen. Die häufigste Anwendung der Emissionsmessung mit der Flamme ist die Bestimmung von Alkalimetallen im Bereich der pharmazeutischen Analytik. Die Methode ist sehr empfindlich und einfach in der Durchführung.
Zugunsten einer preiswerteren Analytik (keine Lampe notwendig) verzichtet man auf wesentliche Vorteile einer Messung in Atomabsorption (bessere Linearität, weiterer Arbeitsbereich, geringere Abhängigkeit von der Flammentemperatur). Im Pharmabereich existieren teilweise 30 Jahre alte Vorschriften, die eine Messung in Emission vorschreiben, obwohl sich in allen anderen Bereichen inzwischen die Atomabsorption durchgesetzt hat.
Aufbau
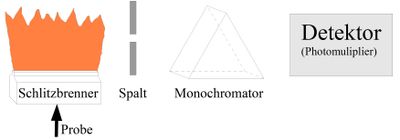
ICP-OES (Optische Emission)
ICP-OES steht für englisch Inductively Coupled Plasma Optical Emission Spectrometry, also der optischen Emissionsspektrometrie mittels induktiv gekoppeltem Plasma. Das "A" in der älteren Bezeichnung ICP-AES steht für Atomic, was jedoch etwas irreführend ist, da in der OES Ionenlinien eine dominante Rolle spielen und nicht Atomlinien.
Die Methode des induktiv gekoppelten Plasmas beruht auf der Verwendung eines sehr heißen (ca. 10000 K) Argon-Plasmas zur Anregung der optischen Emission der zu analysierenden Elemente. Die Grundlagen erarbeiteten unabhängig voneinander Greenfield und Fassel 1964/65. Das erste kommerzielle Gerät wurde 1975 vorgestellt. Die ICP-OES Technik ist inzwischen in der Umweltanalytik, Materialforschung, Metall- oder Pharmaindustrie sehr weit verbreitet.
Prinzip
Ein Plasma ist ein ionisiertes Gas, das neben Atomen auch Elektronen und Ionen enthält. Als Gas wird aufgrund seiner gegenüber den zu bestimmenden Elementen großen Ionisierungsenergie (15,76 eV), seiner chemischen Inertheit, seines vergleichsweise niedrigen Preises, sowie der fehlenden Bandenspektren, meist Argon (einatomiges Gas) verwendet. Die Energieübertragung erfolgt dabei nach der Zündung durch einen Teslafunken durch das in den Spulen anliegende Hochfrequenzfeld. Freie Elektronen werden nun durch das anliegende Feld beschleunigt und heizen durch Kollision mit den Atomrümpfen das Plasma auf. Bedingt durch die hohe Teilchendichte im Plasma erhitzen sich Plasma und Probenaerosol auf 6000–12.000 K (abhängig von der RF-Leistung des Hochfrequenzgenerators in Watt). Die herrschenden Temperaturen sind dabei lokal unterschiedlich, man unterscheidet Ionisations-, Elektronen- und Anregungstemperaturen. Wichtig ist vor allem die Anregungstemperatur mit ca. 6000 K. Das Probenaerosol wird dabei durch die Mitte des Plasmastromes geleitet, ohne dessen Stabilität/Gleichgewicht zu beeinflussen.
Aufbau
Die wichtigsten Teile eines ICP-Spektrometers sind Hochfrequenzgenerator (27 MHz oder 40 MHz), Plasmafackel, Probenzerstäuber (Ultraschall, 1 MHz) und das eigentliche Spektrometer. Der Monochromator moderner OES ist hauptsächlich in der Echelle-Anordnung aufgebaut, da bei dieser Technik aufgrund der kontinuierlichen Emission des Spektrums eine wesentlich bessere Auflösung als in der AAS notwendig ist. Am häufigsten wird ein Polychromator verwendet, da mit ihm die simultane Messung vieler Elemente in kurzer Zeit und sehr stabil möglich ist. Meist kommen Echelle-Polychromatoren in Verbindung mit einem CCD-Flächendetektor zum Einsatz. Die Eigenschaften des Argonplasmas lassen sich in dieser Kombination am besten nutzen:
- Großer dynamischer Messbereich
- Multielementtechnik
- Gute Langzeitstabilität für große Messserien
Die elektromagnetischen Wellen können an zwei verschiedenen Stellen von der Plasmafackel aufgenommen werden. 1. axial, d. h. vom Ende der Fackel (verlängerte Achse) und 2. radial, d. h. von der Seite.
MPT-AES (Mikrowellen-Plasmafackel-AES)
MPT-AES steht für engl. microwave plasma torch atomic emission spectrometry
Es ist ein Verfahren der Spurenanalytik und dient zur empfindlichen Elementanalyse. Der Vorteil bei diesem Verfahren liegt im relativ einfachen Aufbau und in der Verwendung von preiswerten Komponenten.
Insbesondere der Gasverbrauch ist im Vergleich zur ICP-OES bedeutend geringer.Interferenzen in der MPT-AES
Auch hier unterscheidet man spektrale Störungen und nicht-Spektrale Störungen. Chemische Störungen sind kaum von Bedeutung, da die meisten chemischen Verbindungen durch die hohen Temperaturen in der Induktionszone (10.000-12.000 K) des Plasmas dissoziiert werden. Die spektralen Störungen entstehen durch Emissionslinien der Fremdelemente (Interferenten) und Moleküle in der Probenmatrix. Hierzu zählen:
- Direkte Überlagerung von Linien
- Kontinuumsstrahlung aus der Matrix
- Emission von Molekülbanden, wie: -OH, C2, CN, NO, N2
Diese Störungen lassen sich durch:
- geeignete Untergrundanpassung
- die Betrachtung mehrerer Linien pro Element
- spektrale Entfaltung der Linie durch Messung Blindlösung/Analyt/Störer
- Inter-Element-Korrektur
- Standardadditionsverfahren
- veränderte Beobachtungsrichtung
beseitigen.
Zu den nicht-spektralen Störungen zählen auch hier die physikalischen Eigenschaften der Probenlösungen, wie:
- Dichte
- Oberflächenspannung
- Viskosität
Diese können die Zerstäubungseigenschaften, Zerstäuberkammeraerodynamik und den Probentransport nachhaltig beeinflussen. Weiterhin zählen zu den nicht-spektralen Störungen Veränderungen der Anregungsbedingungen im Plasma durch:
- Temperaturänderungen
- Änderungen der Elektronenanzahl im Plasma (Impedanz)
Sie lassen sich durch geeignete Matrixanpassungen, sowie Standard-Additionsverfahren beseitigen.
AFS (Atomfluoreszenzspektroskopie)
Sowohl Fluoreszenz als auch Phosphoreszenz sind Formen der Lumineszenz (kaltes Leuchten). Fluoreszenz ist jedoch dadurch gekennzeichnet, dass sie nach dem Ende der Bestrahlung rasch (meist innerhalb einer Millionstel Sekunde) endet. Bei der Phosphoreszenz hingegen kommt es zu einem Nachleuchten, das von Sekundenbruchteilen bis hin zu Stunden dauern kann.
Atomfluoreszenz ist die optische Emission von in Gasphase gebrachten Atomen, die durch Absorption von elektromagnetischer Strahlung (z. B. Photonen) in einen höheren Energiezustand gebracht wurden. Der Hauptvorteil der Fluoreszenzspektroskopie gegenüber der AAS ist die größere Sensibilität die durch das geringere Hintergrundrauschen ermöglicht wird. Die resonante Anregungsmethode bietet eine selektive Anregung des Analyten um Interferenzen zu vermeiden. Die AFS ermöglicht eine Untersuchung der elektronischen Struktur der Atome und quantitative Messungen. Eine Analyse von Lösungen oder Feststoffen setzt voraus, dass der Analyt aufgelöst, verdampft und atomisiert wird. Das erfolgt in einer Heat-Pipe oder in einem Flamm- oder Graphitofen es sollte auf relativ niedrigen Temperaturen erfolgen. Die eigentliche Anregung der Atome erfolgt durch eine Hohlkathodenlampe (HKL) oder durch einen Laser. Die Detektion erfolgt ähnlich der der Atomemissionsspektroskopie mittels Monochromatoren und Photomultiplier.
ICP-MS (Massenspektrometrie)
siehe Hauptartikel ICP-MS
ICP-MS steht für Inductively Coupled Plasma – Mass Spectrometry, also der Massenspektrometrie mittels induktiv gekoppeltem Plasma.
Im Gegensatz zu den vorherigen Techniken wird in der ICP-MS kein durch Atome absorbiertes oder emittiertes Licht beobachtet, sondern es wird der Einschlag von Ionen bzw. deren Massen auf einen Detektor gemessen. Eine flüssige Probe wird über eine Pumpe angesaugt, in einem Zerstäuber zerstäubt und in einem Argonplasma in der sog. Fackel oder Torch zerschlagen und ionisiert. Die zumeist einfach geladenen Ionen werden im Hochvakuum mit Hilfe einer elektrischen Linsenoptik fokussiert, in einem Quadrupol nach ihrem Masse/Ladungs-Verhältnis aufgetrennt und treffen dann auf einen Detektor, der die Anzahl der Ionen pro Masse aufzeichnet und damit eine quantitative Analyse der Elemente ermöglicht. Die ICP-MS kombiniert die Fähigkeit einer Multielementanalyse sowie den weiten linearen Arbeitsbereich aus der ICP-OES mit den sehr guten Nachweisgrenzen der Graphitrohr-AAS und übertrifft sie sogar. Außerdem ist sie eine der wenigen Analysetechniken, die die Quantifizierung von Isotopenkonzentrationen bei der Analyse der Elemente erlaubt. Allerdings können wegen des Engpasses am Übergang vom Argon-Plasma zum Hochvakuum nur begrenzte Salzlasten eingebracht werden. Teure hochauflösende Geräte (Sektorfeldgeräte mit Auflösungen bis ca. 10.000) können Atom-/Molekülmassen im Nachkommastellen-Bereich unterscheiden; sie trennen somit molekulare Interferenzen von den eigentlich zu messenden Elementmassen. Ihre Empfindlichkeit ist gegenüber den Quadrupolgeräten nochmals um bis zum Faktor 1000 gesteigert. Sie eignen sich wegen ihres geringen Substanzbedarfs daher auch hervorragend zur Analyse radioaktiver Proben.
Anwendung / Fazit / Nachweisgrenzen
Nachweisgrenzen NGr (3 mal Untergrundrauschen) Methode in ppt* ICP/OES pneumat. Zerstäuber >30 ICP/OES Ultraschall ca. >10 Graphitrohrofen/AAS >0,1 ICP/MS >0,02 *Diese NGr können nur unter optimalen Bedingungen erreicht werden (1 ppt = 1E−12 g/g = 1 pg/g = 1 ng / kg oder 1 Teil auf 1 000 000 000 000 Teile)
(Anmerkung: Es sei auf die sprachlich abweichende Bedeutung zwischen deutschem- und englischsprachlichen Raum hingewiesen: billion = Milliarde, trillion = Billion usw.)
Vorteil der ICP-OES gegenüber der Flammen-AAS ist eine erheblich höhere Temperatur des Plasmas gegenüber der Flamme (10.000 K gegenüber 2800 K). Dadurch wird nicht nur der Atomisierungsgrad erhöht (Boltzmann-Verteilung), sondern die angeregten Atome der zu bestimmenden Elemente werden zusätzlich ionisiert. Dies hat wiederum einen entscheidenden Vorteil gegenüber der AAS, da Ionenlinien im Gegensatz zu Atomlinien bei hohen Temperaturen recht unempfindlich gegenüber Anregungsstörungen sind. Außerdem erreicht man eine längere Verweilzeit und eine bessere Temperaturhomogenität, Präzision/Reproduzierbarkeit und Nachweisgrenzen. Eine simultane Multielementanalyse von bis zu 70 Elementen ist heute Stand der Technik. Einsatz findet die ICP-OES heute überwiegend in der Spuren- und Wasseranalytik. Im Umweltbereich werden mittels ICP-OES Böden, Pflanzen, Abfälle und Düngemittel auf die Elementzusammensetzung gemessen. ICP-OES eignet sich hervorragend zur Analyse auch stärker radioaktiver Proben[1], da keine störende Radioaktivität in den Analysatorteil (Optik, Halbleiter-Chip, Verstärker) eingebracht wird, es wird lediglich das emittierte Licht analysiert (anders als z. B. bei MS) und eine nahezu 100%-ige Absaugung der Plasma-Gase ist heutzutage (ab ca. 2000) Stand der Technik.
Einzelnachweise
Literatur
- Bernhard Welz, Michael Sperling: Atomabsorptionsspektrometrie. 4., neubearbeitete Auflage, Weinheim 1999, ISBN 3-527-28305-6
- D. A. Skoog, J. J. Leary: Instrumentelle Analytik. Springer-Lehrbuch, Berlin 1996, ISBN 3-540-60450-2
- D. C. Harris: Quantitative Chemical Analysis 7. Auflage, W. H. Freeman and Company, New York 2003, ISBN 0-7167-7694-4
- K. Cammann: Instrumentelle Analytische Chemie. Spektrum Akademischer Verlag 2000, ISBN 3-8274-0057-0
- J.-M. Mermet, E. Poussel: ICP Emission Spectrometers: Analytical Figures of Merit. Applied Spectroscopy, 1995, 49, 12A
- J. Nölte: ICP Emissionsspektrometrie für Praktiker. Grundlagen, Methodenentwicklung, Anwendungsbeispiele. Verlag Wiley-VCH, Weinheim 2002, ISBN 3-527-30351-0
- G. Wünsch: Optische Analysenverfahren zur Bestimmung anorganischer Stoffe. Sammlung Göschen Bd. 2606, Verlag de Gruyter, Berlin, ISBN 3-11-003908-7
Weblinks
- Principle of Atomic Emission
- R.D. Beaty; J.D. Kerber "Concepts, Instrumentation and Techniques in Atomic Absorption Spectrophotometry", Second Edition The PerkinElmer Corporation (96-seitiges Buch zur AAS als pdf-Dokument (422kb) in englischer Sprache (an der Eingabemaske nach "Beaty Kerber" suchen))
- Doktorarbeit mit Informationen vor allem zur OES, Autor: Ulrich Engel, Universität Dortmund, Anno 2000 (PDF)
- "The 30-Minute Guide to ICP-MS", (8-seitige PerkinElmer Applikationsnote als pdf-Dokument (370kb) in englischer Sprache (an der Eingabemaske nach "30-minute guide" suchen))
- Grundsatzuntersuchungen mit CS-AAS








Wikimedia Foundation.