- Molekularphysik
-
In der Molekülphysik wird die Struktur und das Verhalten von Molekülen untersucht. Sie baut auf Erkenntnissen der Atomphysik und Quantenmechanik auf. Ein wichtiges Modell zu Berechnung der Moleküleigenschaften ist die Born-Oppenheimer-Näherung.
Eine wichtige Methode der Molekülphysik ist die Spektroskopie, in der nicht nur die elektronischen Energiezustände, sondern auch Schwingungs- und Rotationszustände auftreten.
Das in der Atomphysik wichtige Orbitalmodell wird in der Molekülphysik um Molekülorbitale erweitert.
Die Molekülphysik kann als Grenzgebiet zwischen Physik und Chemie aufgefasst werden. Untersuchungsobjekte und -methoden entsprechen weitgehend denen der physikalischen Chemie.
Inhaltsverzeichnis
Molekülspektren
In Molekülen werden die von den Atomen bekannten elektronischen Energieniveaus von Schwingungs- und diese von Rotationszuständen weiter unterteilt. Die Energieabstände zwischen elektronischen Zuständen sind am größten und liegen bei einigen Elektronenvolt, die zugehörige Strahlung liegt etwa im sichtbaren Bereich. Die Strahlung von Schwingungsübergängen liegt im mittleren Infrarot ungefähr zwischen 3 und 10 µm, die von Rotationsübergängen im fernen Infrarot, ca. zwischen 30 und 150 µm. Das Spektrum eines Moleküls besteht in der Regel aus viel mehr Linien als das eines Atoms. Zu einer Änderung des elektronischen Zustandes gehört ein sogenanntes Bandensystem, wobei jede einzelne Bande einem gleichzeitigen Schwingungsübergang beim elektronischen Übergang entspricht. Jede Bande besteht wiederum aus einzelnen Spektrallinien, zu denen jeweils ein parallel zum elektronischen Übergang und zum Schwingungsübergang stattfindender Rotationsübergang gehört.
Hier werden vor allem zweiatomige Moleküle betrachtet, bei denen die Zustände einfacher dargestellt werden können.
Rotation eines zweiatomigen Moleküls
Näherungsweise (für niedrige Rotationsquantenzahlen, d. h. wenn das Molekül nicht so schnell rotiert, dass der Kernabstand merklich steigt) kann man das Molekül als starr betrachten, d. h. der Abstand zwischen den Atomkernen ist konstant. Es kommt zur Quantisierung des Drehimpulses (
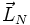 genannt, obwohl die zugehörige Quantenzahl J genannt wird):
genannt, obwohl die zugehörige Quantenzahl J genannt wird):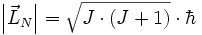
mit den Rotationsquantenzahlen J = 0,1,2,... und der vom Planckschen Wirkungsquantum abgeleiteten Größe
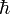 . Die Rotationsenergie ist
. Die Rotationsenergie ist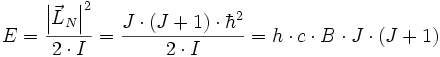
mit dem Trägheitsmoment I und der sogenannten Rotationskonstante
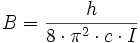
des Moleküls. Durch spektroskopische Messungen kann man die Rotationskonstante bestimmen und so auf das Trägheitsmoment und den Kernabstand schließen.
Der Abstand zwischen den Energieniveaus J = n und J = n + 1 beträgt
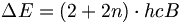 , steigt also mit steigender Quantenzahl. Die Auswahlregel für Übergänge mit Absorption oder Emission eines Photons ist
, steigt also mit steigender Quantenzahl. Die Auswahlregel für Übergänge mit Absorption oder Emission eines Photons ist 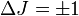 , zusätzlich muss das Molekül ein permanentes Dipolmoment besitzen, was bei Molekülen mit zwei gleichartigen Atomen nicht der Fall ist; deshalb haben diese Moleküle kein reines Rotationsspektrum.
, zusätzlich muss das Molekül ein permanentes Dipolmoment besitzen, was bei Molekülen mit zwei gleichartigen Atomen nicht der Fall ist; deshalb haben diese Moleküle kein reines Rotationsspektrum.Die Energieunterschiede zwischen Rotationsniveaus liegen im Bereich der typischen thermischen Energien von Teilchen bei Zimmertemperatur. Im thermischen Gleichgewicht sind die Energiezustände nach der Boltzmann-Statistik besetzt. Dabei muss man aber beachten, dass es sich bei dem Zustand mit der Quantenzahl J eigentlich um 2J + 1 entartete Zustände (mit den Richtungsquantenzahlen
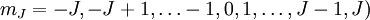 handelt. Die Besetzungsdichte ist daher proportional zu
handelt. Die Besetzungsdichte ist daher proportional zu 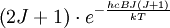 . Das Rotationsspektrum ist außerdem noch von den Übergangswahrscheinlichkeiten zwischen den Zuständen abhängig. Wenn diese ungefähr gleich sind, dann spiegeln die Intensitäten der Spektrallinien die Besetzungsdichten wieder. Typischerweise steigt die Besetzungsdichte von J = 0 weg mit steigendem J anfangs durch den Faktor 2J + 1 bis zu einem Maximum, um dann durch den exponentiellen Faktor wieder abzufallen; so sehen dann auch oft Rotationsspektren aus. Die Abstände zwischen den Spektrallinien sind dabei alle gleich, weil die Abstände zwischen den Energieniveaus bei einer Erhöhung von J um 1 immer um 2hcB steigen und durch die Auswahlregel nur Übergänge zum nächstgelegenem Niveau möglich sind.
. Das Rotationsspektrum ist außerdem noch von den Übergangswahrscheinlichkeiten zwischen den Zuständen abhängig. Wenn diese ungefähr gleich sind, dann spiegeln die Intensitäten der Spektrallinien die Besetzungsdichten wieder. Typischerweise steigt die Besetzungsdichte von J = 0 weg mit steigendem J anfangs durch den Faktor 2J + 1 bis zu einem Maximum, um dann durch den exponentiellen Faktor wieder abzufallen; so sehen dann auch oft Rotationsspektren aus. Die Abstände zwischen den Spektrallinien sind dabei alle gleich, weil die Abstände zwischen den Energieniveaus bei einer Erhöhung von J um 1 immer um 2hcB steigen und durch die Auswahlregel nur Übergänge zum nächstgelegenem Niveau möglich sind.Schwingungen eines zweiatomigen Moleküls
Die Atome eines zweiatomigen, hantelförmigen, Moleküls können auch gegeneinander schwingen. Die einfachste Näherung ist hier eine harmonische Schwingung; die potentielle Energie eines Atoms muss hier mit der Entfernung von einem Gleichgewichtsabstand r0 zum anderen Atom quadratisch ansteigen. Die Energieniveaus eines quantenmechanischen harmonischen Oszillators (Quantenzahl ν) sind äquidistant:
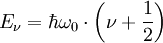
Reale Moleküle weichen jedoch stark von diesem Verhalten ab, das Potenzial ist nicht harmonisch (anharmonischer Oszillator) und steigt bei Annäherung an das andere Atom viel stärker an als bei Entfernung - hier nähert es sich asymptotisch der Dissoziationsenergie Edis des Moleküls. Eine bessere Näherung als das harmonische Potenzial ist das sogenannte Morse-Potential. Diese Funktion bildet das reale Potenzial viel besser ab und die Schrödinger-Gleichung ist trotzdem noch analytisch lösbar. Für verschwindend kleine Abstände geht sie nicht gegen unendlich:
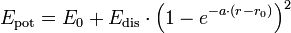
E0 ist die Nullpunktsenergie des Potenzials, a ein Parameter:
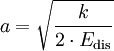
k ist hier die Federkonstante des am besten passenden harmonischen Potenzials.
Die Energieniveaus können dann mit Hilfe der Schrödingergleichung berechnet werden:
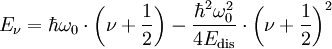
Im Gegensatz zum harmonischen Oszillator liegen nun die erlaubten benachbarten Schwingungszustände nicht mehr äquidistant, sondern verringern ihren Abstand näherungsweise mit 1 / ν2. Damit ist das Korrespondenzprinzip erfüllt: Bei der Annäherung an die Dissoziationsenergie liegen die quantenmechanisch erlaubten Zustände praktisch unendlich dicht.
Die Auswahlregeln für Übergänge zwischen Schwingungsniveaus sind
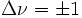 für den harmonischen Oszillator, für den anharmonischen Oszillator sind auch
für den harmonischen Oszillator, für den anharmonischen Oszillator sind auch 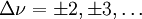 mit abnehmender Wahrscheinlichkeit erlaubt. Man beobachtet praktisch nie reine Schwingungsspektren, weil diese durch Rotationsspektren überlagert sind.
mit abnehmender Wahrscheinlichkeit erlaubt. Man beobachtet praktisch nie reine Schwingungsspektren, weil diese durch Rotationsspektren überlagert sind.Rotations-Schwingungs-Wechselwirkung
 Schwingungs-Rotationsübergänge und resultierendes Spektrum
Schwingungs-Rotationsübergänge und resultierendes SpektrumWeil das Trägheitsmoment des Moleküls durch die Schwingungen schwankt, muss man zur Energie des Moleküls bei genauerer Betrachtung noch die Rotations-Schwingungs-Wechselwirkungsenergie addieren. Nach den folgenden Ansatz für die Gesamtenergie
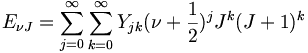
kann man die sogenannten Dunham-Koeffizienten Yjk den experimentellen Resultaten anpassen.
Das effektive Potenzial für die Molekülschwingung im zweiatomigen Molekül wird durch die Rotation erhöht (Epot,0 ist das Potenzial ohne Rotation):
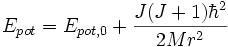
Dadurch kommt es bei höheren Rotationsquantenzahlen zur Ausbildung einer sogenannten Rotationsbarriere: Bei steigender Entfernung der Atomkerne wächst das effektive Potenzial von einem Minimum (Gleichgewichtslage) zu einem Maximum (der Rotationsbarriere), das bereits über der Dissoziationsenergie liegt, um danach wieder zur Dissoziationsenergie abzufallen. Dadurch kann sich das Molekül in einem Schwingungszustand "hinter" der Rotationsbarriere befinden, dessen Energie höher ist als die Dissoziationsenergie. Es kann dann zur Dissoziation durch den Tunneleffekt kommen. Bei sehr hohen Rotationsquantenzahlen wird auch das Potenzialminimum über die Dissoziationsenergie gehoben, bei noch höheren Rotationsquantenzahlen gibt es schließlich kein Minimum und somit keine stabilen Zustände mehr.
Elektronische Zustände im zweiatomigen Molekül
Ähnlich wie bei Atomen wird auch hier der Zustand eines Elektrons durch eine Hauptquantenzahl n und eine Bahndrehimpulsquantenzahl l angegeben, wobei den verschiedenen Werten von l wie beim Atom Buchstaben zugeordnet sind (s, p, d, f, ...). Das elektrische Feld ist aber nicht mehr kugelsymmetrisch, deshalb muss sich der Bahndrehimpuls relativ zur Kernverbindungsachse einstellen. Die Projektion des Bahndrehimpulses
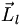 auf die Kernverbindungsachse nennt man
auf die Kernverbindungsachse nennt man 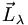 mit der zugehörigen Quantenzahl λ. Für die unterschiedlichen Werte von λ schreibt man griechische Buchstaben, die den römischen Buchstaben bei den Werten von l entsprechen (σ, π, δ, φ, ...). Man schreibt dann z. B. den Grundzustand des Wasserstoffmoleküls (1sσ)2: 2 Elektronen befinden sich im Grundzustand n = 1, l = 0, λ = 0.
mit der zugehörigen Quantenzahl λ. Für die unterschiedlichen Werte von λ schreibt man griechische Buchstaben, die den römischen Buchstaben bei den Werten von l entsprechen (σ, π, δ, φ, ...). Man schreibt dann z. B. den Grundzustand des Wasserstoffmoleküls (1sσ)2: 2 Elektronen befinden sich im Grundzustand n = 1, l = 0, λ = 0.Die Kopplung der einzelnen Drehimpulse zu einem Moleküldrehimpuls erfolgt zweckmäßig je nach den Stärken der Wechselwirkungen in unterschiedlicher Reihenfolge, man spricht von den Hundschen Kopplungsfällen (a) - (e) (nach Friedrich Hund).
Die Summe der auf die Kernverbindungsachse projizierten Bahndrehimpulse nennt man
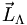 mit der Quantenzahl Λ; die Summe der Elektroneneigendrehimpulse (Spins) nennt man wie beim Atom
mit der Quantenzahl Λ; die Summe der Elektroneneigendrehimpulse (Spins) nennt man wie beim Atom 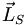 mit der Quantenzahl S; die Projektion dieses Gesamtspins auf die Kernverbindungsachse nennt man
mit der Quantenzahl S; die Projektion dieses Gesamtspins auf die Kernverbindungsachse nennt man 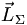 mit der Quantenzahl Σ; die Summe von und nennt man
mit der Quantenzahl Σ; die Summe von und nennt man 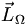 mit der Quantenzahl Ω. Der mechanischen Drehimpuls des Moleküls tritt mit den elektronischen Drehimpulsen auch noch in Wechselwirkung.
mit der Quantenzahl Ω. Der mechanischen Drehimpuls des Moleküls tritt mit den elektronischen Drehimpulsen auch noch in Wechselwirkung.Für die elektronischen Zustände werden oft auch andere Bezeichnungen verwendet: X steht für den Grundzustand, A, B, C, ... stehen für die immer höher angeregten Zustände (kleine Buchstaben a, b, c, ... kennzeichnen in der Regel Triplettzustände).
Hamiltonoperator für Moleküle
Es ist üblich, den Hamiltonoperator nicht in SI-Einheiten, sondern in sogenannten atomaren Einheiten zu schreiben, da dies die folgenden Vorteile birgt:
- Da Naturkonstanten nicht mehr explizit auftauchen, sind die Ergebnisse in atomaren Einheiten einfacher hinzuschreiben und unabhängig von der Genauigkeit der involvierten Naturkonstanten. Die in atomaren Einheiten berechneten Größen lassen sich dennoch einfach in SI-Einheiten zurückrechnen.
- Numerische Lösungsverfahren der Schrödingergleichung verhalten sich angenehmer, da die zu verarbeitenden Zahlen wesentlich näher bei der Zahl 1 liegen, als dies in SI-Einheiten der Fall ist.
Der Hamiltonoperator ergibt sich zu
- H = Te + Tk + Vee + Vkk + Vek
mit
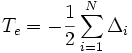 , der kinetischen Energie der Elektronen
, der kinetischen Energie der Elektronen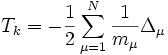 , der kinetischen Energie der Atomkerne
, der kinetischen Energie der Atomkerne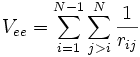 , der potentiellen Energie der Wechselwirkung zwischen den Elektronen
, der potentiellen Energie der Wechselwirkung zwischen den Elektronen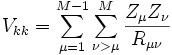 , der potentiellen Energie der Wechselwirkung zwischen den Kernen
, der potentiellen Energie der Wechselwirkung zwischen den Kernen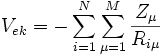 , der potentiellen Energie der Wechselwirkung zwischen den Elektronen und Atomkernen.
, der potentiellen Energie der Wechselwirkung zwischen den Elektronen und Atomkernen.
Hierbei sind i und j die Indizes über die Elektronen, μ bzw. ν die Indizes über die Atomkerne, rij der Abstand zwischen dem i-ten und dem j-ten Elektron, Rμν der Abstand zwischen dem μ-ten und dem ν-ten Atomkern und Riμ der Abstand zwischen dem i-ten Elektron und dem μ-ten Atomkern, Zμ die Kernladungszahl des μ-ten Atomkerns.
Die zeitunabhängige Schrödingergleichung ergibt sich dann zu HΨ = EΨ, wobei allerdings in der Praxis die Gesamtschrödingergleichung mit Hilfe der Born-Oppenheimer-Näherungen in eine elektronische Schrödingergleichung (mit festen Kernkoordinaten) und eine Kernschrödingergleichung aufgeteilt wird. Die Lösung der Kernschrödingergleichung setzt dabei die Lösung der elektronischen Schrödingergleichung für alle (relevanten) Kerngeometrien voraus, da die elektronische Energie als Funktion der Kerngeometrie dort eingeht. Die elektronische Schrödingergleichung ergibt sich formal durch setzen von Tk = 0.
Siehe auch
Literatur
- Hermann Haken, Hans-Christoph Wolf: Molekülphysik und Quantenchemie: Einführung in die experimentellen und theoretischen Grundlagen. Springer 2006, ISBN 978-3-540-30315-2

Wikimedia Foundation.