- NSIP
-
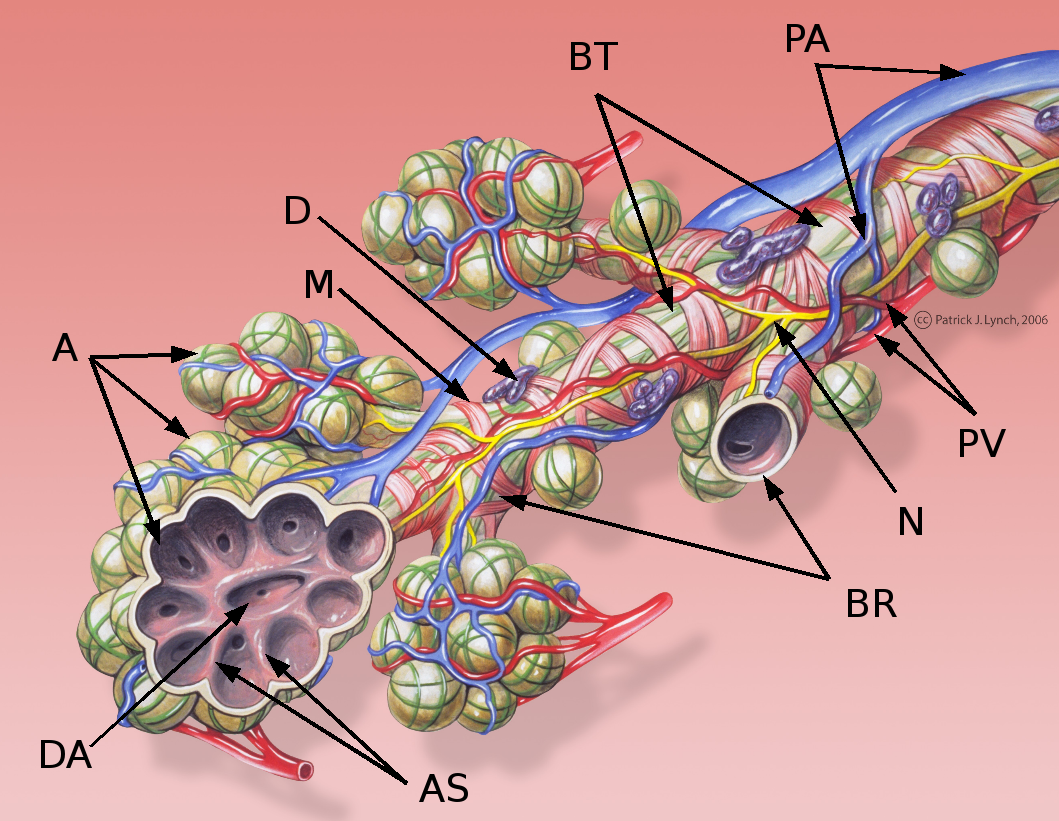
Klassifikation nach ICD-10 J84 Sonstige interstitielle Lungenkrankheiten ICD-10 online (WHO-Version 2006) 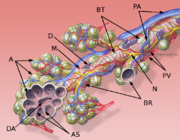 Schematische Darstellung eines Bronchiolus terminalis
Schematische Darstellung eines Bronchiolus terminalis
A: Alveolen
AS: Alveolarsepten
BR: Bronchioli respiratorii
BT: Bronchiolus terminalis
D: Schleimhautdrüse
DA: Ductus alveolaris
M: zirkuläre Muskelschicht des Bronchiolus
N: Nerv
PA: Äste der Pulmonalarterie
PV: Äste der PulmonalvenenDie idiopathischen interstitiellen Pneumonien (IIP) bilden eine Gruppe von seltenen Krankheitsformen, die durch ein unterschiedliches Ausmaß von Lungenentzündung (Pneumonie) und -vernarbung (Lungenfibrose), hauptsächlich des Bindegewebes der Lunge (Interstitium), gekennzeichnet sind. Sie unterscheiden sich hinsichtlich der Krankheitszeichen (der Symptome), der Befunde vor allem aus bildgebenden Untersuchungsverfahren, der Pathologie, des Krankheitsverlaufs und der Behandlungsmöglichkeiten.[1] Die Ursache für die Entstehung dieser Krankheitsbilder ist nicht bekannt – sie werden daher als „idiopathisch“ bezeichnet.[2] Die IIP gehören zur großen Gruppe der interstitiellen Lungenerkrankungen (englisch: Interstitial Lung Disease oder ILD), in der mehr als 200 eigenständige Krankheitsbilder klassifiziert werden. Das Leitsymptom der interstitiellen Lungenerkrankungen ist Atemnot (Dyspnoe).
Bei allen IIP-Formen kommt es zu einer Lungenentzündung unterschiedlichen Schweregrads, die in erster Linie das Bindegewebe (Interstitium) der Lunge betrifft. Darüber hinaus kann es zu einer Vernarbung der Lunge kommen, die als Lungenfibrose bezeichnet wird. Bei einigen Formen steht die Lungenfibrose im Vordergrund und die Entzündung ist nur eine Begleitreaktion. Die Prognose oder der Krankheitsverlauf und die Behandlungsmöglichkeiten sind unter anderem davon abhängig, welchen Stellenwert die Entzündung oder die Fibrose bei der entsprechenden Krankheitsform einnimmt.[3]
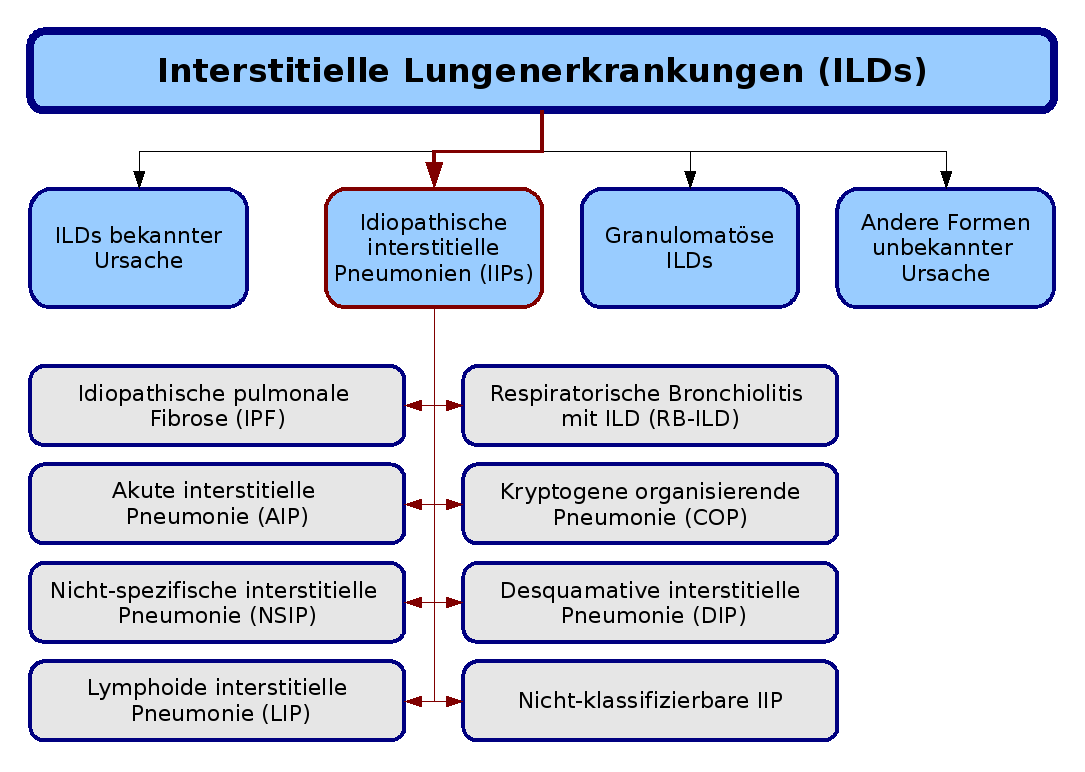
Klassifikation
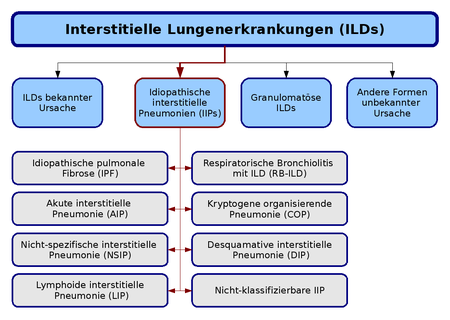 Die Klassifikation der interstitiellen idiopathischen Pneumonien (rot hervorgehoben) im Kontext der interstitiellen Lungenerkrankungen, basierend auf der Konsensus-Klassifikation der American Thoracic Society und der European Respiratory Society von 2002.
Die Klassifikation der interstitiellen idiopathischen Pneumonien (rot hervorgehoben) im Kontext der interstitiellen Lungenerkrankungen, basierend auf der Konsensus-Klassifikation der American Thoracic Society und der European Respiratory Society von 2002.Die aktuelle Klassifikation der idiopathischen interstitiellen Pneumonien (IIP) basiert auf einer Richtlinie, die gemeinsam von der American Thoracic Society (ATS) und der European Respiratory Society (ERS) im Jahre 2002 veröffentlicht wurde. Nach dieser Klassifikation werden sieben Krankheitsformen unterschieden, die jeweils durch unterschiedliche klinische, radiologische und pathologische Kriterien definiert sind. Darüber hinaus wurde eine Kategorie für nicht klassifizierbare IIP eingeführt, der alle Fälle zugeordnet werden, bei denen keine klare Abgrenzung möglich ist:
- idiopathische pulmonale Fibrose (IPF, idiopathische Lungenfibrose),
- nicht spezifische interstitielle Pneumonie (NSIP),
- kryptogene organisierende Pneumonie (COP),
- akute interstitielle Pneumonie (AIP),
- respiratorische Bronchiolitis mit interstitieller Lungenerkrankung (RB-ILD),
- desquamative interstitielle Pneumonie (DIP) und
- lymphoide interstitielle Pneumonie (LIP).[1]
Die Bedeutung der neuen Klassifikation liegt vor allem in der fachgebietsübergreifenden (interdisziplinären) Definition der verschiedenen Formen, die in zurückliegenden Veröffentlichungen von Pathologen, Radiologen und Internisten überwiegend unabhängig voneinander vorgenommen wurde. Es wurde klargestellt, dass das histopathologische Muster von der klinischen Diagnose abgegrenzt werden muss. Eine sichere klinische Diagnose beruht sowohl auf klinischen, radiologischen als auch auf histopathologischen Befunden. Zwar ist der Evidenzgrad der Klassifikation gering, da sie auf Erkenntnissen von Experten beruht, sie bildet aber die Grundlage für zukünftige Studien, da es jetzt einheitliche Kriterien auf diesem Gebiet gibt.[4]
Epidemiologie
Die epidemiologischen Daten zu den idiopathischen interstitiellen Pneumonien sind nicht sehr zuverlässig. Amerikanische Arbeiten zu der übergeordneten Gruppe aller interstitiellen Lungenerkrankungen (ILD) wurden unter anderem in New Mexiko durchgeführt. Nach dieser Studie beträgt die Prävalenz der ILD 80 Fälle pro 100.000 Einwohner bei Männern und 67 Fälle pro 100.000 Einwohner bei Frauen. Die Diagnose Lungenfibrose und idiopathische Lungenfibrose (IPF) macht etwa 45 Prozent aller Fälle von ILD aus. [5] Die Prävalenz der IPF beträgt nach dieser Studie 20 Fälle pro 100.000 Einwohner bei Männern und 13 Fälle pro 100.000 Einwohner bei Frauen. Da diese Studie vor Veröffentlichung der neuen Klassifikation durchgeführt wurde, ist die Aussagekraft dieser Angaben fragwürdig. Eine neuere Studie geht von einer höheren Prävalenz aus. [6] Es gibt Hinweise dafür, dass die IPF in westlichen Ländern zunimmt. [7] Die Epidemiologie der anderen IIP-Formen ist noch weniger gut erforscht. [1]
Anatomische Grundlagen
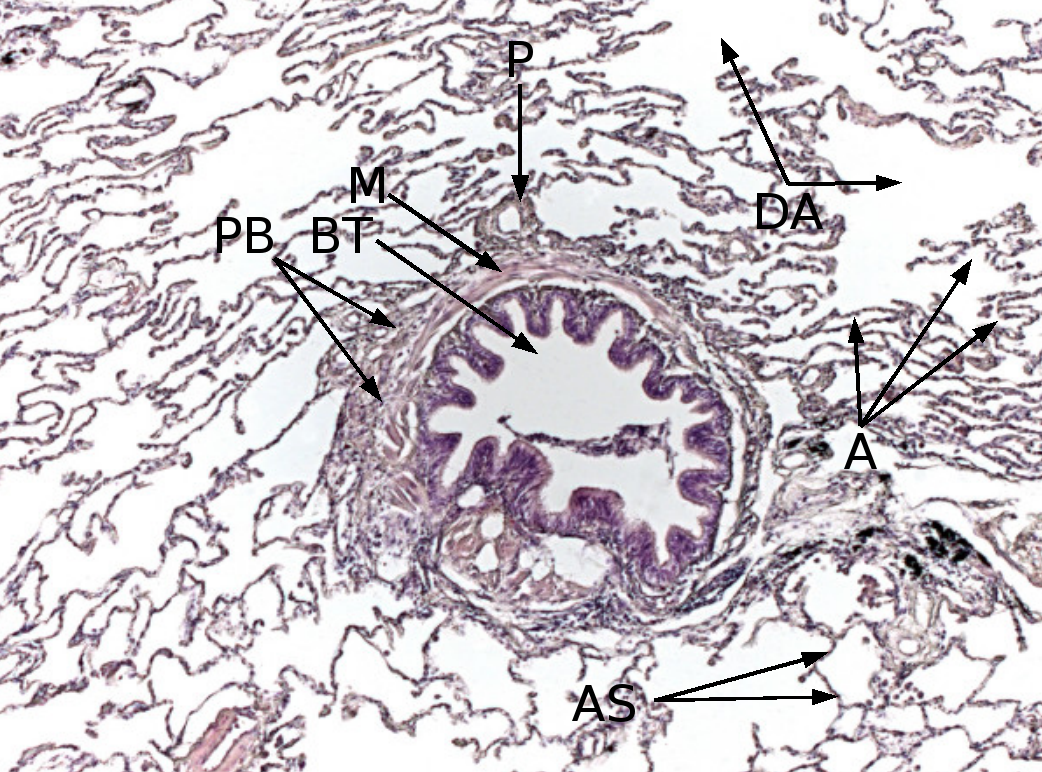
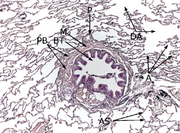 Histologie des Lungengewebes
Histologie des Lungengewebes
PB: Peribronchiales Bindegewebe, BT: Bronchiolus terminalis, M: zirkuläre Muskelschicht des Bronchiolus, P: Ast der Pulmonalarterie, DA: Ductus alveolares, A: Alveolen, AS: Alveolarsepten.
Färbung mit Hämatoxylin-Eosin (HE).Das Interstitium (Zwischengewebe) der Lunge befindet sich in Form der so genannten Interalveolarsepten zwischen den Wänden der Lungenbläschen (Alveolen). Letztere stellen, da in ihnen der Gasaustausch zwischen Blut und Luft erfolgt, das eigentliche funktionelle Lungengewebe (Parenchym) dar. Interstitium und Parenchym arbeiten jedoch bei der Lungenmechanik eng zusammen und bilden eine funktionelle Einheit. Das Interstitium ist das Grundgerüst der Lunge und hält die Architektur der Lungenbläschen und damit der Blut-Luft-Schranke während des Atmungszyklus' aufrecht. Der Bindegewebsanteil der Interalveolarsepten besteht aus Zellen, vor allem den Bindegewebsbildnern (Fibroblasten), den von ihnen gebildeten Strukturproteinen und Substanzen wie Kollagen, elastische Fasern, Proteoglykane sowie anderen Glycoproteinen.
Den Hauptteil des Interstitiums bilden Kollagenfasern, die bis zu 20 Prozent der Lungentrockenmasse ausmachen. Im Interstitium der Lunge herrscht der Kollagentyp I vor, dem vermutlich die Hauptrolle bei der Aufrechterhaltung der Form und Dehnbarkeit der Lungenbläschen zukommt. Typ IV ist nur in den Basalmembranen der Lungenbläschen und Blutgefäße vorhanden. Kollagen vom Typ III und V ist nur in geringem Umfang (5 bis 10 Prozent) im Lungengewebe ausgebildet.[8] Die elastischen Fasern bilden ein verzweigtes Netzwerk im Interstitium der Lunge. Sie sind die treibende Kraft der Ausatmung (Exspiration).
Proteoglykane bilden die Grundsubstanz des Interstitiums, sie sorgen für die Aufrechterhaltung des räumlichen Aufbaus des Lungengewebes. Ihre genaue Bedeutung bei der Lungenfunktion ist nicht im Einzelnen bekannt. In der Lunge finden sich Hyaluronsäure, Chondroitinsulfat A und C, Dermatansulfat, Heparin und Heparinsulfat. Zu den weiteren Glycoproteinen gehören Fibronectin und Laminin, die vor allem in den Basalmembranen vorkommen.
Pathogenese
Als Pathogenese werden die Mechanismen bezeichnet, die zur Entstehung einer Erkrankung beitragen. Bei den idiopathischen interstitiellen Pneumonien (IIP) spielen vor allem die Entzündung und die Fibrose des Lungengewebes die entscheidende Rolle. Sie haben bei den verschiedenen Formen einen unterschiedlichen Stellenwert. Die Pathogenese ist aber nur unvollständig geklärt.
Entzündung
Bei den meisten interstitiellen Lungenerkrankungen und den meisten Formen der idiopathischen interstitiellen Pneumonien scheint die Entzündungsreaktion die entscheidende Rolle in der Pathogenese zu spielen. Bei einer mikroskopischen Untersuchung werden bei diesen Erkrankungen, vor allem im Anfangsstadium, große Ansammlungen von Entzündungszellen (Makrophagen, Granulozyten und Lymphozyten) im Lungengewebe gefunden. Fibroseareale treten nur in geringem Umfang auf und nur selten entwickelt sich eine Lungenfibrose im Endstadium. Die Lungenfibrose ist dann eine Folge der Entzündungsreaktion. Diese Theorie wird dadurch bestärkt, dass die Erkrankungen auf entzündungshemmende und immunsuppressive Therapien gut ansprechen. Durch Beseitigung der Entzündung als Ursache für die Lungenfibrose kann ein weiteres Fortschreiten verhindert werden.
Eine Sonderstellung nehmen vor allem die idiopathische pulmonale Fibrose und wahrscheinlich auch die akute interstitielle Pneumonie ein. Die Hypothese, dass eine Entzündungsreaktion die Bedingung für die Entstehung einer Lungenfibrose ist, scheint hier nicht aufrecht zu halten zu sein. Die Entzündungsreaktion spielt hier offensichtlich eine untergeordnete Rolle oder ist nur eine Begleitreaktion der Lungenfibrose.[9][10].
Lungenfibrose
Die Lungenfibrose ist eine Reaktion, bei der es zur Vernarbung des Lungengewebes kommt. Für die Entstehung der Lungenfibrose spielen verschiedene Zelltypen eine wichtige Rolle. Die wichtigsten sind nach heutigem Kenntnisstand Fibroblasten, Endothelzellen und Alveolarepithelzellen. Nach einer Schädigung des Lungengewebes, deren Ursache bei den idiopathischen interstitiellen Pneumonien unbekannt ist, kommt es entweder zunächst zu einer Entzündung und anschließend zu einer Aktivierung der Fibroblasten, oder sie werden direkt, das heißt ohne vorausgehende Entzündung aktiviert. Dies ist davon abhängig welches Modell zugrunde gelegt wird. Die Aktivierung der Fibroblasten wird durch verschiedene Botenstoffe initiiert, zum Beispiel Wachstumsfaktoren wie Transforming growth factor beta 1 (TGF-β1) und verschiedene Interleukine, die aus anderen Zellen und den Fibroblasten selbst freigesetzt werden. Dies führt zu einer Zellvermehrung (Proliferation) der Fibroblasten sowie zu einer Neubildung von Grundsubstanz und Bindegewebsfasern. Normalerweise wird diese Reaktion streng reguliert, das heißt, dass nach Beseitigung der Schädigung oder der Wundheilung die Aktivität der Fibroblasten gehemmt wird: einerseits durch die geringere Ausschüttung von Botenstoffen, andererseits durch den programmierten Zelltod (Apoptose) der Fibroblasten.[11] Bei der Lungenfibrose funktionieren diese Regulationsmechanismen nicht adäquat. Dies führt dazu, dass zu viel Bindegewebe gebildet wird. Die Alveolarsepten werden breiter, so dass es zu einer Gasaustausch- oder Diffusionsstörung (respiratorische Insuffizienz) kommt. Darüber hinaus verliert die Lunge ihre Dehnbarkeit (Compliance) und kann sich im Rahmen der Einatmung nur durch erhöhte Atemarbeit im ausreichenden Maße ausdehnen. Es entsteht eine sogenannte restriktive Ventilationsstörung. Beide Faktoren – die Gasaustauschstörung und die restriktive Ventilationsstörung – führen zum Leitsymptom der Lungenfibrose, der Atemnot. In fortgeschrittenen Stadien der Lungenfibrose wandern die Fibroblasten darüber hinaus in das Lumen der Alveolen ein und bilden auch dort neues Bindegewebe. Es bilden sich fibrotische Areale in den Alveolen, die als fibrotische Foci bezeichnet werden. Dadurch wird die Struktur der Lunge in diesen Bereichen zerstört, was mit einem vollständigen Funktionsverlust einhergeht.[12]
Symptome
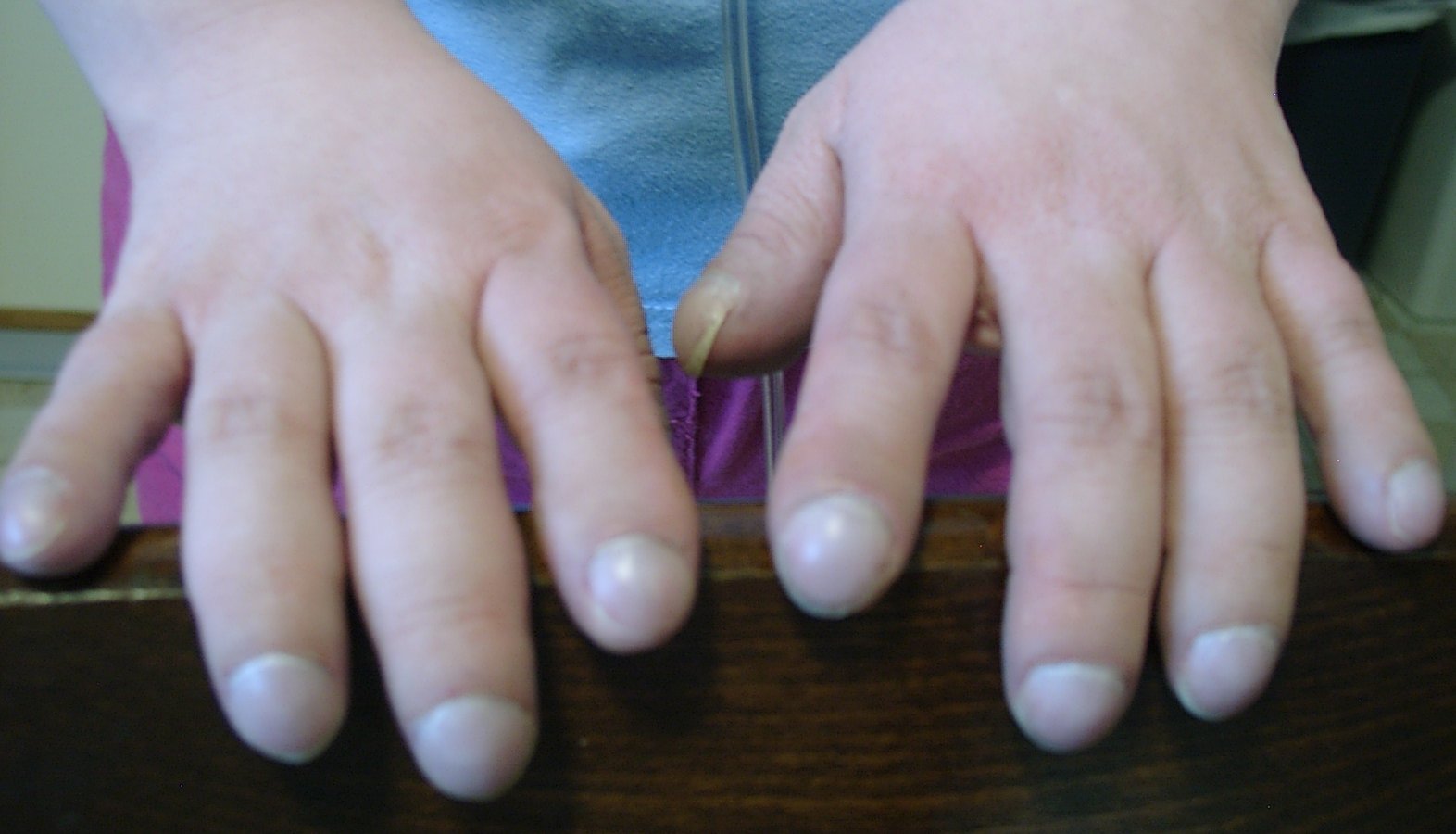
 Trommelschlägelfinger und Uhrglasnägel sind klinische Zeichen eines Sauerstoffmangels (Hypoxämie). Sie können im Rahmen von idiopathischen interstitiellen Pneumonien vorkommen.
Trommelschlägelfinger und Uhrglasnägel sind klinische Zeichen eines Sauerstoffmangels (Hypoxämie). Sie können im Rahmen von idiopathischen interstitiellen Pneumonien vorkommen.Das Leitsymptom der idiopathischen interstitiellen Pneumonien, wie auch der meisten anderen interstitiellen Lungenerkrankungen ist eine Atemnot, die im Anfangsstadium nur bei körperlicher Belastung auftritt. Da es sich bei den IIP typischerweise um eine restriktive Ventilationsstörung handelt, haben die Betroffenen vor allem Schwierigkeiten bei der Einatmung. Bei fortgeschrittenem Schweregrad (Progredienz) der Erkrankung kann die Atemnot, je nach Krankheitsform, auch in Ruhe auftreten und im Endstadium können die Erkrankungen in eine respiratorische Insuffizienz übergehen. Charakteristisch für Lungenfibrosen ist auch ein trockener Husten ohne Auswurf.
Aufgrund der eingeschränkten Funktionsfähigkeit der Lunge, hinsichtlich des Gasaustausches und der Compliance, kann es zu einem Sauerstoffmangel im Blut, einer so genannten Hypoxämie kommen. Diese kann sich in Form einer Zyanose, das heißt einer violetten bis bläulichen Verfärbung der Haut, der Schleimhäute und der Fingernägel bemerkbar machen. Auch Trommelschlägelfinger und Uhrglasnägel können sich als Zeichen der Hypoxämie ausbilden. Bei starkem Sauerstoffmangel können auch Bewusstseinsstörungen auftreten.[13]
Diagnose
Basisdiagnostik
Anamnese und körperliche Untersuchung
Am Beginn der Untersuchung steht die Erhebung einer ausführlichen Krankheitsvorgeschichte (Anamnese). Dabei werden die Patienten unter anderem nach den aktuellen Beschwerden, bereits bekannten Erkrankungen, eingenommenen Medikamenten und Erkrankungen in der Verwandtschaft befragt. Die Anamnese hat einen hohen Stellenwert, da sie dabei hilft, andere Erkrankungen auszuschließen und eine Verdachtsdiagnose zu stellen. Bei vielen Erkrankungen werden vom Patienten charakteristische Symptome geschildert, so dass sich die Verdachtsdiagnose häufig, nach weiteren Untersuchungen, als die Richtige herausstellt.
Bei Lungenerkrankungen kann der im deutschsprachigen Raum verbreitete Frankfurter Bogen hilfreich sein.[14] Dieser Bogen wird vom Patienten ausgefüllt und berücksichtigt nahezu alle wichtigen Gesichtspunkte, die bei Lungenerkrankungen von Bedeutung sind. Die Symptome der idiopathischen interstitiellen Pneumonien sind unspezifisch. Die Atemnot, die bei allen IIP-Formen vorkommt, weist darauf hin, dass eine Lungenerkrankung vorliegen könnte, sie kann aber auch durch zahlreiche andere Erkrankungen hervorgerufen werden, wie zum Beispiel durch Herzerkrankungen. Dies gilt in ähnlicher Weise auch für die anderen geschilderten Symptome. Die Diagnose oder die Verdachtsdiagnose einer IIP kann daher nicht allein aufgrund der Anamnese gestellt werden.
Auf die Anamnese folgt üblicherweise eine ausführliche körperliche Untersuchung. Der Schwerpunkt liegt bei Lungenerkrankungen auf dem Betrachten (Inspektion) der gesamten Körperoberfläche, dem Abklopfen (Perkussion) des Brustkorbs sowie dem Abhören (Auskultation) der Lunge mit einem Stethoskop. Es sind vor allem die Fibrose-typischen Veränderungen, die bei einer IIP mit einer körperlichen Untersuchung aufgedeckt werden können. Häufig kann man auskultatorisch ein deutliches Knisterrasseln hören, das auch als Sklerophonie bezeichnet wird und durch feinblasige Nebengeräusche gekennzeichnet ist, die gegen Ende der Ein- und Ausatmung auftreten. Mit Hilfe der Perkussion können die ebenfalls für Lungenfibrose typischen hochstehenden Lungengrenzen aufgedeckt werden. Die bereits genannten Symptome Zyanose, Trommelschlägelfinger und Uhrglasnägel fallen bei der Inspektion auf. Die körperliche Untersuchung lässt bei IIP ebenfalls keine sichere Diagnose zu, kann das Feld der möglichen Erkrankungen aber weiter einschränken.[15]
Technische Untersuchungsverfahren
Zur Basisdiagnostik bei einem Verdacht auf eine ILD oder eine IIP gehören technische Untersuchungsverfahren, insbesondere die Untersuchung der Lungenfunktion sowie eine Röntgenaufnahme des Brustkorbs. Bei der Bewertung der Lungenfunktion spielt vor allem die Bodyplethysmografie eine wichtige Rolle. Bei einer Lungenfibrose kommt es zu einer restriktiven Ventilationsstörung, die mit dieser Methode aufgedeckt werden kann. In Frühstadien der Erkrankung kann der Nachweis einer solchen Störung allerdings fehlen. Eine Blutgasanalyse vor und nach körperlicher Belastung sowie eine Kohlenmonoxid-Diffusionsmessung können bereits in Frühstadien Hinweise auf das Vorliegen einer IIP liefern. Beide Untersuchungsverfahren gehören ebenfalls zum Repertoire der Basisdiagnostik.[15]
Fibrotische Veränderungen können des Weiteren auf Röntgenbildern der Lunge sichtbar gemacht werden. Auch hier gilt, dass Veränderungen in Frühstadien so gering sein können, dass sie sich der konventionellen Röntgendiagnostik entziehen. Typische Zeichen einer Lungenfibrose im Röntgenbild sind Konsolidierungen, Traktionsbronchiektasien und Milchglasverschattungen. Konsolidierungen entstehen durch fibrotisches Gewebe in den Alveolen und stellen sich im Röntgenbild als helle Areale dar, da sie elektronendichter sind als luftgefüllte Alveolen. Traktionsbronchiektasien sind Ausweitungen der Bronchien und werden durch den Zug verursacht, der auf sie ausgeübt wird und bei einer Lungenfibrose durch die Schrumpfung des Lungengewebes entsteht. Bei den Milchglasverschattungen handelt es sich ebenfalls um helle Areale, die durch die Zerstörung der Lungenarchitektur verursacht werden.[16]
Weiterführende Diagnostik
Besteht nach der Basisdiagnostik der dringende Verdacht auf eine IIP, wird in der Regel eine spezielle Computertomografie (CT), die als High Resolution CT bezeichnet wird, durchgeführt. Im Vergleich zur normalen CT wird hiermit eine höhere Auflösung (engl. high resolution) erreicht. Mit der HRCT können die meisten Fälle von IIP mit hoher Wahrscheinlichkeit identifiziert werden und die verschiedenen Krankheitsformen differenziert werden. Typische Merkmale der Lungenfibrose im CT sind neben Milchglasverschattungen und Traktionsbronchiektasien eine so genannte Honigwabenlunge oder kurz Honigwaben (engl. honeycombing). Die Honigwabenlunge wird so aufgrund ihrer ähnlichen Struktur zu den Bienenwaben bezeichnet. Die Honigwaben entstehen infolge der Degeneration des Lungengewebes.[16]
Wenn eine Diagnose oder eine Differenzierung mit dieser Methode nicht möglich ist, muss auf invasive Untersuchungsverfahren zurückgegriffen werden. An erster Stelle der invasiven Untersuchungen steht die Bronchoskopie mit bronchoalveolärer Lavage (BAL) sowie die transbronchiale Biopsie. Ist auch hiermit kein Ausschluss oder eine Sicherung der Diagnose möglich, muss eine offene Lungenbiopsie durchgeführt werden, die den Goldstandard in der Diagnostik der IIP darstellt. Nach den Empfehlungen der Konsensus-Konferenz sollten mindestens drei Proben aus verschiedenen Lungenlappen mit einer Mindestgröße von jeweils 2 cm entnommen werden.[15]
Therapie
Pharmakologische Therapie
Die Therapie der idiopathischen interstitiellen Pneumonien (IIP) wird im Wesentlichen mit zwei Gruppen von Medikamenten durchgeführt. Zum einen mit einem Glukokortikoid, etwa Prednisolon, und zum anderen mit so genannten Immunsuppressiva, vor allem Azathioprin und Cyclophosphamid. Die Wirksamkeit der Medikamente ist abhängig von der Form der IIP, an welcher der Patient erkrankt ist. Während die pharmakologische Therapie bei der idiopathischen Lungenfibrose (IPF) und der akuten interstitiellen Pneumonie (AIP) keinen oder nur einen vorübergehenden Effekt auf den Verlauf der Erkrankung hat, sprechen die anderen Formen gut auf eine entzündungshemmende Therapie an, so dass eine Heilung erfolgen oder zumindest die mittlere Überlebenszeit erheblich verlängert werden kann.
Das unterschiedliche Ansprechen auf die entzündungshemmende Therapie ist wahrscheinlich auf eine unterschiedliche Pathogenese der verschiedenen Formen zurückzuführen. Bei der IPF und AIP steht nach aktuellem Forschungsstand die Fibrose im Vordergrund und die Entzündungsreaktion ist nur eine Begleitreaktion oder eine Folge davon. Bei den anderen Formen ist es dagegen die Entzündung des Lungeninterstitiums, die das Krankheitsgeschehen bestimmt und die erst im Endstadium der Erkrankung durch Fibrosevorgänge überlagert wird.
Für Therapie der IPF und AIP werden daher große Hoffnungen in Substanzen gelegt, die das Fortschreiten der Fibrose, das heißt die Zellvermehrung der Fibroblasten und die Kollagensynthese, hemmen können. Eine Vielzahl solcher Substanzen befindet sich zu Zeit (2008) in klinischen Studien. Sie sind aber für die Standardtherapie der IIP bisher nicht zugelassen. [17]
Lungentransplantation
Eine Lungentransplantation kann in Erwägung gezogen werden, wenn sich die IIP oder die Lungenfibrose im Endstadium befindet und nicht auf eine medikamentöse Therapie anspricht. Die Lungentransplantation erhöht bei fortgeschrittenen Lungenfibrosen die Überlebensrate.[18] Die Transplantation eines Lungenflügels ist der Transplantation von zwei Lungenflügeln ebenbürtig. [19] Die Indikation für eine Lungentransplantation wird häufig nicht rechtzeitig gestellt. Nach einer Studie, in die 1376 Patienten einbezogen wurden, starben mehr als 30 Prozent aufgrund langer Wartezeiten, noch bevor die Lungentransplantation durchgeführt werden konnte.[20]
Klinische Unterschiede der idiopathischen interstitiellen Pneumonien
Idiopathische pulmonale Fibrose (IPF)
.JPG) Histologisches Muster einer Usual Interstitial Pneumonia (UIP, gewöhnliche interstitielle Pneumonie). Sichtbar sind die Fibroseareale in den Interalveolarsepten. Färbung mit Hämatoxylin-Eosin (HE).
Histologisches Muster einer Usual Interstitial Pneumonia (UIP, gewöhnliche interstitielle Pneumonie). Sichtbar sind die Fibroseareale in den Interalveolarsepten. Färbung mit Hämatoxylin-Eosin (HE).Die idiopathische pulmonale Fibrose oder idiopathische Lungenfibrose ist die häufigste IIP und hat die zweitschlechteste Prognose. Die durchschnittliche Überlebenszeit beträgt nach Diagnosestellung durchschnittlich drei Jahre und die Fünfjahres-Überlebensrate liegt zwischen 20 und 40 Prozent. Die Patienten sind zum Zeitpunkt der Diagnosestellung meist älter als 60 Jahre und Männer sind etwas häufiger betroffen als Frauen.[21]
Die morphologischen Veränderungen sind überwiegend in Bereich der Lungenbasis, das heißt im unteren Bereich der Lunge, lokalisiert. Das typische histologische Muster der IPF wird als Usual Interstial Pneumonia (UIP, gewöhnliche interstitielle Pneumonie) bezeichnet und ist charakterisiert durch eine fleckenförmige Fibrose sowie eine honigwabenartige Struktur der Lunge. Das Muster der UIP ist charakteristisch aber nicht spezifisch für die IPF und kommt auch bei anderen interstitiellen Lungenerkrankungen vor. Im Röntgenbild zeigt sich in fortgeschrittenen Stadien der Erkrankung eine basale retikuläre (netzförmige) Gefäßzeichnung. In der HRCT (High Resolution Computertomographie) sind Traktionsbronchiektasien sowie Honigwaben typisch. Häufig lassen sich auch Milchglasverschattungen nachweisen.
Der Krankheitsbeginn der IPF ist schleichend und durch eine zunehmende Atemnot gekennzeichnet. Die IPF spricht auf eine Therapie mit Glukokortikoiden, auch in Kombination mit immunsuppressiven Medikamenten, in der Regel nicht an, so dass die zurzeit einzige wirksame therapeutische Maßnahme die Lungentransplantation ist.
Nicht spezifische interstitielle Pneumonie (NSIP)
Die nicht spezifische interstitielle Pneumonie (NSIP) ist unter den idiopathischen interstitiellen Pneumonien (IIP) die zweithäufigste und variantenreichste Form. Die Betroffenen sind meist zwischen 50 und 60 Jahre alt und damit im Durchschnitt etwas jünger als an der IPF erkrankte Patienten. Frauen und Männer sind gleichhäufig betroffen.
Die NSIP zeigt bezüglich der morphologischen Veränderungen keine typische Lokalisation. Die Veränderungen sind vielmehr über die gesamte Lunge homogen und meist symmetrisch verteilt. Das histologische Muster wird ebenfalls als NSIP bezeichnet. Es existieren zwei Untergruppen (Subtypen) der NSIP, ein zellulärer und ein fibrotischer Subtyp. Beim zellulären Subtyp wird das histologische Erscheinungsbild von Entzündungszellen dominiert, beim fibrotischen Subtyp sind zusätzlich ausgeprägte Fibroseareale vorhanden. Wie das histologische Bild ist auch das radiologische Bild variantenreich. Neben diffus verteilten Milchglastrübungen findet man Mikronoduli und Honigwaben. In fortgeschrittenen Stadien ist die normale Lungenstruktur im Röntgen- und im CT-Bild nur noch schwer zu erkennen, da sie durch Bronchiektasien, Zysten und Konsolidierungen zerstört ist.
Die Symptomatik der NSIP unterscheidet sich nur unwesentlich von der bei IPF. Sie zeigt allerdings einen milderen Verlauf, ist langsamer fortschreitend und hat eine bessere Prognose. Die Prognose ist außerdem abhängig vom histologischen Subtyp und ist beim zellulären Subtyp besser als beim fibrotischen Subtyp, da dieser besser auf eine antientzündliche Therapie anspricht. Insgesamt ist die medikamentöse Therapie wesentlich erfolgsversprechender als dies bei der IPF der Fall ist. Da die morphologischen Veränderungen der NSIP so vielseitig sind, bereitet die Diagnose den Ärzten, im Vergleich mit anderen IIP, die größten Schwierigkeiten. Dies gilt insbesondere für den fibrotischen Subtyp.[22]
Kryptogene organisierende Pneumonie (COP)
Als kryptogene organisierende Pneumonie wird ein Krankheitsbild bezeichnet, dessen klinische, radiologische und pathologische Eigenschaften relativ charakteristisch sind. Wie bei der NSIP gibt es bei der COP keine Bevorzugung eines Geschlechts und das durchschnittliche Alter der Patienten liegt zwischen 50 und 60 Jahren. Nichtraucher sind in etwa doppelt so häufig betroffen wie Zigarettenraucher. Die Ursache für diese statistische Auffälligkeit ist nicht bekannt.
Das histologische Muster wird als organisierende Pneumonie (OP) bezeichnet. Es ist gekennzeichnet durch das Auftreten von Granulationsgewebe, das die Bronchiolen und Alveolen ausfüllt. Das Parenchym der Lunge wird durch diese Veränderungen nicht zerstört. Dieses Muster tritt nur selten in der idiopathischen Form auf. Häufig können andere Erkrankungen als sekundäre Ursache für diese Veränderungen verantwortlich gemacht werden, zum Beispiel Kollagenosen sowie infektiös- oder medikamentös-induzierte Erkrankungen. Eine ähnliche Histologie tritt auch bei der Bronchiolitis obliterans auf. Daher wurde die COP früher auch als Bronchiolitis-obliterans-organisierende Pneumonie (BOOP) bezeichnet. Um Verwechslungen mit dem eigenständigen Krankheitsbild der Bronchiolitis obliterans zu vermeiden, ist diese Bezeichnung inzwischen nicht mehr gebräuchlich. Auf Röntgen- und CT-Bildern stellt sich das Granulationsgewebe in Form von so genannten Konsolidierungsarealen dar, die vor allem im Bereich um die Bronchien herum (peribronchial) sowie direkt unter der Pleura (subpleural) lokalisiert sind. Um die Konsolidierungen können Milchglasverschattungen auftreten.
Das Krankheitsbild beginnt meist mit allgemeinen Symptomen, wie Unwohlsein, Fieber, Gewichtsverlust und trockenem Husten. Die Symptome nehmen innerhalb von ein bis drei Monaten zu. Die COP lässt sich gut mit Glukokortikoiden behandeln und besitzt eine gute Prognose. Häufig flammt die Erkrankung nach Absetzen der Therapie wieder auf. Dies wird als Rezidiv bezeichnet. Beim Auftreten von Rezidiven kann die Erkrankung erneut mit Glukokortikoiden behandelt werden.
Akute interstitielle Pneumonie (AIP)
Die akute interstitielle Pneumonie (AIP) hat, im Gegensatz zu den anderen idiopathischen interstitiellen Pneumonien (IIP), einen akuten Verlauf. Sie ist durch das plötzliche Auftreten von Symptomen gekennzeichnet und mündet häufig in eine fortschreitende Störung der Atmung (Ateminsuffizienz). Die Erkrankung hat die schlechteste Prognose der IIP. In über der Hälfte der Fälle ist der Verlauf tödlich. Die meisten Todesfälle treten ein bis zwei Monate nach Krankheitsbeginn auf.
Dem Ausbruch der AIP geht häufig eine Infektion der oberen Atemwege voraus, die durch allgemeines Krankheitsgefühl, Gliederschmerzen, Fieber und Schüttelfrost gekennzeichnet sein kann. Bei den Patienten kommt es dann innerhalb weniger Tage zu Atemnot (Dyspnoe), die zunächst nur bei Belastung auftritt. Bereits im Anfangsstadium lässt sich ein Sauerstoffmangel im Blut (Hypoxämie) nachweisen. Charakteristisch ist außerdem ein deutliches Knisterrasseln, das als Zeichen der Konsolidierung im Rahmen einer restriktiven Ventilationsstörung zu werten ist. Aus dem Sauerstoffmangel im Blut (Hypoxämie) entwickelt sich in wenigen Wochen eine Atemnot in Ruhe (Ruhedyspnoe), die in eine Ateminsuffizienz übergehen kann. Der Patient muss in diesem Stadium mit Sauerstoff versorgt und unter Umständen maschinell beatmet werden.
Das histologische Muster wird als diffuse alveoläre Schädigung (engl. diffuse alveolar damage, kurz DAD) bezeichnet und lässt sich nicht von pathologischen Veränderungen unterscheiden, die beim akuten Atemnotsyndrom des Erwachsenen (engl. acute respiratory distress syndrome, kurz ARDS) auftreten. AIP und ARDS unterscheiden sich lediglich in der Ursache. Während das ARDS vor allem im Rahmen einer Sepsis oder eines Schocks auftritt, ist die Ursache der AIP unbekannt. Louis Virgil Hamman und Arnold Rice Rich beschrieben bereits in den 1940er-Jahren ähnliche Fälle, die später als Hamman-Rich-Syndrom zusammengefasst wurden. Wahrscheinlich handelt es sich bei diesen Fällen um die Beschreibung der heute als AIP bezeichneten Erkrankung.
Respiratorische Bronchiolitis mit interstitieller Lungenerkrankung (RB-ILD) und desquamative interstitielle Pneumonie (DIP)
Bei der respiratorischen Bronchiolitis mit interstitieller Lungenerkrankung und der desquamativen interstitielle Pneumonie handelt es sich um Erkrankungen mit ähnlichem, aber unterscheidbarem Erscheinungsbild. Beide Erkrankungen kommen vor allem, die RB-ILD ausschließlich und die DIP in der Regel, bei Rauchern vor. Das mittlere Erkrankungsalter liegt bei jeweils etwa 40-50 Jahren und Männer sind etwa doppelt so häufig betroffen wie Frauen. Die RB-ILD ist die symptomatische Variante der respiratorischen Bronchiolitis, einer Entzündung der Bronchiolen, die bei Rauchern regelmäßig durch Zufall gefunden werden kann. Das histologische Muster der asymptomatischen und der symptomatischen Variante wird gleichfalls als respiratorische Bronchiolitis (RB) bezeichnet.
Die DIP wird von einigen Experten als die fortgeschrittene Variante der RB-ILD angesehen. Dies ist allerdings zweifelhaft, da die DIP im Gegensatz zur RB-ILD gelegentlich auch bei Nichtrauchern vorkommt und aufgrund der doch erheblichen morphologischen Unterschiede (siehe unten). Die Konsensus-Klassifikation sieht aufgrund dieser Unsicherheit eine Trennung beider Varianten vor. Der Begriff der DIP wurde 1969 von Liebow und Carrington unter der falschen Annahme eingeführt, es handele sich bei diesen Zellansammlungen um Abschilferungen des Alveolarepithels. Diese Abschuppung von Zellen wird als Desquamation (von lat. squama „Schuppe“) bezeichnet. Trotzdem wurde der Begriff der DIP in der aktuellen Klassifikation beibehalten, unter anderem, weil es sich bei der DIP um eine Rarität handelt, und nicht durch den besser passenden Begriff Alveolarmakrophagen-Pneumonie ersetzt.[1] Das histologische Muster der DIP wird ebenfalls als DIP bezeichnet.
Das histologische Bild beider Formen ist durch zahlreiche Ansammlungen (Akkumulation) von braun pigmentierten Makrophagen in der Lunge gekennzeichnet. Bei der RB-ILD sind die pigmentierten Makrophagen überwiegend in den Lumina der Bronchiolen (bronchiolozentrisch) lokalisiert, während sie bei der DIP vorwiegend in den Lumina der Alveolen (intraalveolär) zu finden sind. Bei der DIP ist außerdem eine geringgradige Fibrose charakteristisch. Auch bei der Verteilung der morphologischen Veränderungen unterscheiden sich beide Krankheitsbilder. Während die RB-ILD vor allem in den Oberlappen der Lunge zu finden ist, weist die DIP eine bevorzugte subpleurale Lokalisation mit Dominanz in den Unterlappen auf. Dieses Verteilungsmuster ist besonders gut in der HRCT nachweisbar. Bei der RB-ILD sind so genannte zentrilobuläre Knötchen, bei der DIP Milchglasverschattungen charakteristisch.
Die klinische Symptomatik ist durch eine langsame Entwicklung von Atemnot und trockenem Reizhusten gekennzeichnet. Bei etwa der Hälfte der Patienten finden sich außerdem Trommelschlägelfinger. Beide Formen sprechen gut auf Glukokortikoide an und haben eine gute Prognose. Nur in seltenen Fällen entwickelt sich bei der DIP eine respiratorische Insuffizienz, die eine hohe Letalität aufweist.
Lymphoide interstitielle Pneumonie (LIP)
Die LIP tritt bevorzugt bei Frauen mit einem Altersgipfel um das 50. Lebensjahr auf. In der idiopathischen Form ist die LIP die seltenste IIP. Weit häufiger tritt sie sekundär im Rahmen von Autoimmunerkrankungen und bei Immunschwäche auf.
Das histologische Muster wird wie die klinische Diagnose als LIP bezeichnet und ist gekennzeichnet durch Infiltration der Alveolarsepten mit Lymphozyten und Makrophagen sowie durch die peribronchiale Bildung von Lymphfollikeln. Die Veränderungen sind diffus über die ganze Lunge verteilt. In der HRCT sind Milchglasverschattungen charakteristisch.
Die Patienten haben nur eine gering ausgeprägte Symptomatik und leiden vor allem unter leichter Atemnot und Husten. Bei den sekundären Formen stehen die Symptome der Grunderkrankung im Vordergrund. Die LIP kann in seltenen Fällen in eine Lungenfibrose oder in ein malignes Lymphom übergehen. Abgesehen von dieser Komplikation hat die LIP eine gute Prognose. Die Therapie erfolgt in der Regel mit Glukokortikoiden, deren Wirksamkeit bei dieser IIP-Form allerdings noch nicht durch randomisierte Studien bewiesen wurde.
Idiopathische interstitielle Pneumonien bei Tieren
Die Gruppe der idiopathischen interstitiellen Pneumonien bei Tieren ist bislang nur wenig untersucht. Daher gibt auch es kein geeignetes Modelltier für Grundlagenuntersuchungen – die Bleomycin-induzierte Lungenfibrose bei Nagetieren ist nur eine im Erscheinungsbild (phänomenologisch) ähnliche Erkrankung, erfüllt aber nicht die Anforderungen an ein adäquates Modell.[23]
Gehäuft findet man solche chronisch-idiopathischen Erkrankungen bei einigen Terrierrassen (West Highland White Terrier, Staffordshire Bullterrier, Scottish Terrier). Die canine idiopathische Lungenfibrose tritt vor allem bei älteren Tieren auf und entspricht der kryptogenen organisierenden Pneumonie des Menschen.[24] Bei Katzen sind in der Literatur nur wenige Fälle einer IIP beschrieben,[25] nach einer aktuellen Studie entspricht sie bei Katzen der idiopathischen pulmonalen Fibrose (IPF) des Menschen und wird vermutlich durch einen Defekt der Typ-II-Pneumozyten hervorgerufen.[23]
Bei Vögeln gibt es bislang nur einen einzigen Fallbericht bei einer Blaustirnamazone.[26]
Forschungsgeschichte
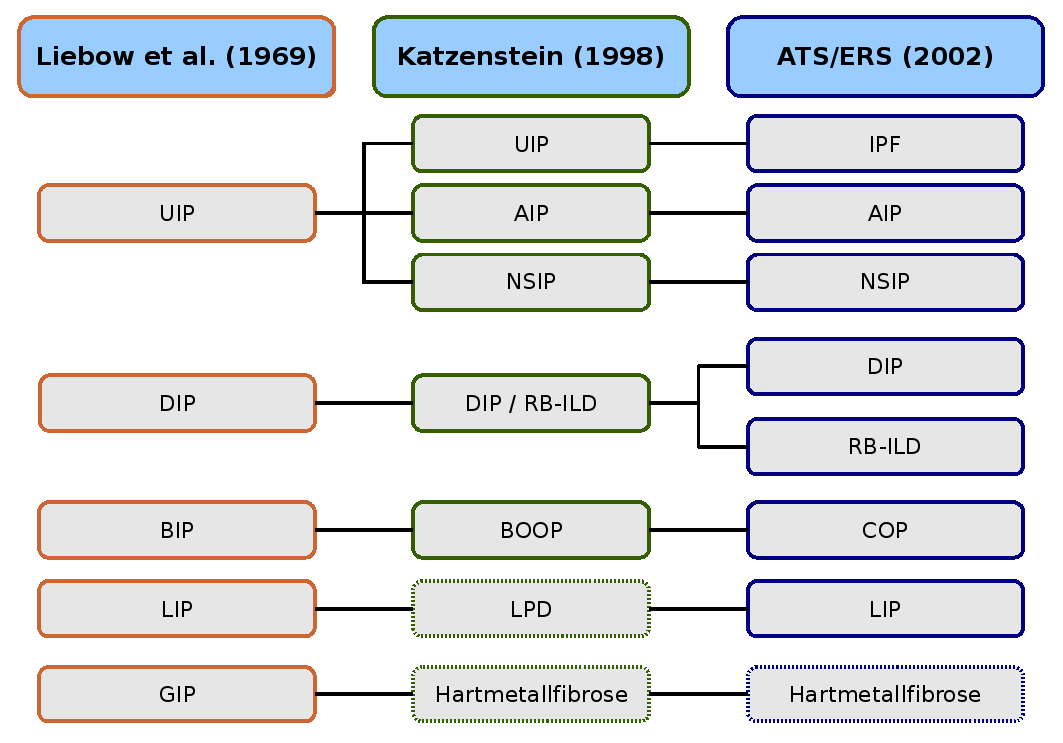
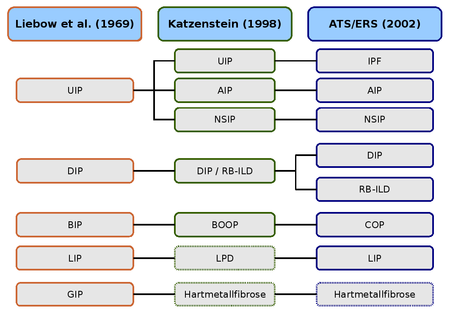 Vergleich der historischen Klassifikationen und der aktuellen Klassifikation der ATS und ERS.
Vergleich der historischen Klassifikationen und der aktuellen Klassifikation der ATS und ERS.
AIP (akute interstitielle Pneumonie),
BIP (bronchiolitische interstitielle Pneumonie),
BOOP (Bronchiolitis obliterans mit organisierender Pneumonie),
COP (kryptogene interstitielle Pneumonie),
DIP (desquamative interstitielle Pneumonie),
GIP (Riesenzell-Pneumonie),
LPD (lymphoproliferative Erkrankungen),
LIP (lymphoide interstitielle Pneumonie),
NSIP (nicht spezifische interstitielle Pneumonie),
RB-ILD (respiratorische Bronchiolitis mit interstit. Lungenerkrankung),
UIP (usual interstitial pneumonia).Die erste Beschreibung einer interstitiellen Lungenerkrankung geht auf das Jahr 1892 zurück und stammt von dem kanadischen Mediziner William Osler. Er erkannte bereits das vielfältige Erscheinungsbild und wies auf die Notwendigkeit und die Schwierigkeit einer weiteren Klassifikation der Veränderungen hin.
Der Internist Louis Virgil Hamman und der Pathologe Arnold Rice Rich berichteten 1944 über vier Patientenfälle, die von einer akuten diffusen interstitiellen Fibrose betroffen waren. Nach ihnen wurde das Hamman-Rich-Syndrom benannt. Dieser Begriff wurde daraufhin eine Zeit lang für alle Erkrankungen verwendet, bei denen es zu einer chronischen diffusen Fibrosierung der Lunge mit unbekannter Ursache kam, obwohl es sich bei den beschriebenen Fällen um akute Verläufe handelte. Wahrscheinlich beschrieben die Autoren seinerzeit die heute als akute interstitielle Pneumonie bezeichnete Form.
Die erste histologische Klassifikation idiopathischer interstitieller Lungenerkrankungen stammt von den beiden Pathologen Averill Abraham Liebow und Charles B. Carrington (1969). Die damalige Klassifikation unterschied fünf histologische Muster, die erstmals unter dem Oberbegriff idiopathische Lungenfibrose geführt wurden. Das Hamman-Rich-Syndrom wurde als akute Variante der „Usual Interstitial Pneumonia“ (UIP, gewöhnliche interstitielle Pneumonie) eingeordnet.
Die Liebow-Carrington-Klassifikation wurde 1997 von Anna-Luise A. Katzenstein und 1998 in Zusammenarbeit mit Jeffrey L. Myers weiterentwickelt und dem aktuellen Forschungsstand angepasst. Die Ursache der „Giant Interstial Pneumonia“ (GCP, Riesenzell-Pneumonie) wurde inzwischen aufgeklärt. Sie wird durch die Inhalation von Metallstäuben ausgelöst und als Hartmetallfibrose bezeichnet. Die lymphoide interstitielle Pneumonie wurde den „lymphoproliferativen Erkrankungen“ (LPD, lymphoproliferative diseases) zugeordnet.[27][28]
Einzelnachweise
- Anmerkung: Der Artikel baut, wenn keine anderen Quellen angegeben wurden, auf der Veröffentlichung der Konsensuskonferenz von 2002 auf: American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med. 2002 Jan 15;165(2):277-304. PMID 11790668
- ↑ a b c d American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med. 2002 Jan 15;165(2):277-304. PMID 11790668
- ↑ Hunninghake GW, Schwarz MI.:State of the art. Does current knowledge explain the pathogenesis of idiopathic pulmonary fibrosis?: a perspective. Proc Am Thorac Soc. 2007 Sep;4(5):449-52. PMID 17684287
- ↑ Leslie KO: Historical perspective: a pathologic approach to the classification of idiopathic interstitial pneumonias. Chest. 2005 Nov;128 (5 Suppl 1):513S-519S. PMID 16304241
- ↑ Costabel U: Idiopathische interstitielle Pneumonien – wozu schon wieder eine Konsensusklassifikation?. Pneumologie 2002;56: 279-280. Online-Version
- ↑ DB Coultas et al.: The epidemiology of interstitial lung diseases. Am. J. Respir. Crit. Care Med., Vol 150, No. 4, 10 1994, 967-972.[1]
- ↑ Raghu G et al.: Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006 Oct 1;174(7):810-6. Epub 2006 Jun 29. PMID 16809633
- ↑ Francis H.Y. Green: Overview of Pulmonary Fibrosis Chest. 2002;122:334S-339S. Free Full Text
- ↑ J.E. Gadek et al.: Role of connective tissue proteases in the pathogenesis of chronic inflammatory lung disease. Environ Health Perspect. 1984 Apr;55:297-306. PMID 6329673
- ↑ King TE: Idiopathic interstitial pneumonias: progress in classification, diagnosis, pathogenesis and management. Trans Am Clin Climatol Assoc. 2004;115:43-78 PMID 17060957
- ↑ Gross et al.: Idiopathic Pulmonary Fibrosis. N Engl J Med 2001 345: 517-525 Online-Version (Zugangsberechtigung erforderlich)
- ↑ Willis BC et al.: Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. 2006 Jun;3(4):377-82 PMID 16738204
- ↑ R. Klinke, S. Silbernagel (Hrsg.): Lehrbuch der Physiologie. 4. Auflage, Thieme-Verlag 2003, ISBN 3-13-796004-5
- ↑ Dietel M et al: Harrisons Innere Medizin. Deutsche Ausgabe der 15. Auflage, ABW Wissenschaftsverlag Berlin 2003, ISBN 3-936072-10-8"
- ↑ Kronenberger H. et al.: Ein neuer Fragebogen zur Erfassung von Lungen- und Atemwegserkrankungen. Prax. Klin. Pneumol. 39:241-251, 1985. Fragebogen als PDF-Version
- ↑ a b c Günther et al.: Klassifikation, Diagnostik und Therapie der idiopathischen interstitiellen Pneumonien: Eine kritische Bestandsaufnahme der gegenwärtig in Deutschland geübten Praxis. Deutsches Ärzteblatt. Ausgabe 24 vom 13. Juni 2003. (html-Version, PDF-Version)
- ↑ a b Kauffmann et al.: Radiologie. 3. Aufl, Urban & Fischer München/Jena 2006, ISBN 3-437-44415-8
- ↑ Costabel U et al.: Lungenfibrosen - Klassifikation, Diagnostik, Therapie. Internist 2003 [Suppl 1]44:S35-S43. Online-Version
- ↑ Hosenpud JD et al.: Effect of diagnosis on survival benefit of lung transplantation for end-stage lung disease. Lancet. 1998 Jan 3;351(9095):24-7. PMID 9433425
- ↑ Meyers BF et al.: Single versus bilateral lung transplantation for idiopathic pulmonary fibrosis: a ten-year institutional experience. J Thorac Cardiovasc Surg. 2000;120:99-107. PMID 10884661
- ↑ De Meester J et al.: Lung transplant waiting list: differential outcome of type of end-stage lung disease, one year after registration. J Heart Lung Transplant. 1999 Jun;18(6):563-71. PMID 10395354
- ↑ Kim DS et al.: Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006 Jun;3(4):285-92. PMID 16738191
- ↑ Monaghan H et al.: Prognostic implications of histologic patterns in multiple surgical lung biopsies from patients with idiopathic interstitial pneumonias. Chest. 2004 Feb;125(2):522-6. PMID 14769733
- ↑ a b K. Williams et al.: Identification of spontaneous feline idiopathic pulmonary fibrosis: morphology and ultrastructural evidence for a type II pneumocyte defect. Chest. 2004 Jun;125(6):2278-2288. PMID 15189952 Volltext
- ↑ B.M. Corcoran et al.: Chronic pulmonary disease in West Highland white terriers. Vet Rec. 1999 May 29;144(22):611-616. PMID 16119057
- ↑ L.A. Cohn et al.: Identification and characterization of an idiopathic pulmonary fibrosis-like condition in cats. J Vet Intern Med. 2004 Sep-Oct;18(5):632-641. PMID 15515577
- ↑ O. Amann et al: Chronic pulmonary interstitial fibrosis in a blue-fronted Amazon parrot (Amazona aestiva aestiva). Avian Dis. 2007 Mar;51(1):150-153. PMID 17461284
- ↑ Katzenstein A, Myers J: Idiopathic Pulmonary Fibrosis - Clinical Relevance of Pathologic Classification. Am J Respir Crit Care Med. 1998 Apr;157(4 Pt 1):1301-15. PMID 9563754
- ↑ Collard R. et al.: Demystifying Idiopathic Interstitial Pneumonia. Arch Intern Med. 2003;163:17-29. PMID 12523913
Literatur
- J. Müller-Quernheim: Interstitielle Lungenerkrankungen. Thieme, Stuttgart 2001, ISBN 3-13-132281-0
- H. Schweisfurth et al.: Wie werden interstitielle Lungenerkrankungen in Deutschland diagnostiziert?. Pneumologie 2003; 57: 373-382. PDF-Datei
Weblinks
- Mikroskopische Abbildungen der sieben verschiedenen Formen der idiopathischen interstitiellen Pneumonien (englisch)
- Hörbeispiel Knisterrasseln mp3-Datei von Lungenärzte im Netz
- Erklärungen zur idiopathischen interstitiellen Pneumonie
Wikimedia Foundation.
Schlagen Sie auch in anderen Wörterbüchern nach:
Idiopathische Lungenfibrose — Klassifikation nach ICD 10 J84 Sonstige interstitielle Lungenkrankheiten … Deutsch Wikipedia
Idiopathische pulmonale Fibrose — Klassifikation nach ICD 10 J84 Sonstige interstitielle Lungenkrankheiten … Deutsch Wikipedia
Idiopathische interstitielle Pneumonie — Klassifikation nach ICD 10 J84 Sonstige interstitielle Lungenkrankheiten … Deutsch Wikipedia
Fibrose pulmonaire — Demande de traduction Idiopathi … Wikipédia en Français
Idiopathic interstitial pneumonia — Classification and external resources Micrograph of usual interstitial pneumonia (UIP). UIP is the most common pattern of idiopathic interstitial pneumonia and usually represents idiopathic pulmonary fibrosis … Wikipedia
Wallops Flight Facility — (WFF) Airport codes|WAL|KWAL, located on the Eastern Shore of Virginia, is operated by the National Aeronautics and Space Administration’s (NASA) Goddard Space Flight Center primarily as a rocket launch site to support science and exploration… … Wikipedia
Idiopathic pulmonary fibrosis — Classification and external resources Extensive lung fibrosis from usual interstitial pneumonitis ICD 10 J … Wikipedia
Hypersensitivity pneumonitis — Classification and external resources Micrograph of hypersensitivity pneumonitis. Lung biopsy. Trichrome stain … Wikipedia
Alliance atlantique — Organisation du traité de l Atlantique Nord Pour les articles homonymes, voir Otan (homonymie). Organisation du traité de l’Atlantique Nord North Atlantic Treaty Organisation (en) OTAN NATO … Wikipédia en Français
Liste des pays de l'OTAN — Organisation du traité de l Atlantique Nord Pour les articles homonymes, voir Otan (homonymie). Organisation du traité de l’Atlantique Nord North Atlantic Treaty Organisation (en) OTAN NATO … Wikipédia en Français
 18+© Academic, 2000-2026
18+© Academic, 2000-2026- Kontaktieren Sie uns: Unterstützung, Werbung
Wörterbücher Export, schritte mit PHP, Joomla, Drupal, WordPress, MODx.
.JPG)