- Plasmazell-Dyskrasie
-
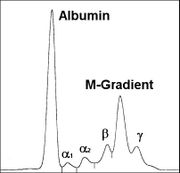 Beispiel einer monoklonalen Gammopathie bei IgA-Plasmozytom
Beispiel einer monoklonalen Gammopathie bei IgA-PlasmozytomKlassifikation nach ICD-10 D47.2 Monoklonale Gammopathie ICD-10 online (WHO-Version 2006) Eine monoklonale Gammopathie ist eine Veränderung der Proteine des Blutplasmas, die mit einer krankhaften Vermehrung eines einzelnen Immunglobulins einhergeht. Sie entstehen bei der Proliferation (Reifung) eines spezifischen Klons von malignen (bösartigen) oder hyperstimulierter Plasmazellen. Der Name beruht auf der Tatsache, dass die Immunglobuline bei der elektrophoretischen Aufteilung der Plasmaproteine (Serumelektrophorese) in der sogenannten γ-Fraktion zu finden sind. Das pathologische Immunglobulin gehört einer der Immunglobinklassen an, am häufigsten ist eine monoklonale Gammopathie vom IgG, IgA oder IgM-Subtyp.
Ursache ist eine Vermehrung eines einzelnen Klons von Antikörper-bildenden Plasmazellen bzw. deren Vorläufern. Aus diesem Grund werden die monoklonalen Gammopathien auch (insbesondere in der englischsprachigen Literatur) als Plasmazell-Dyskrasien (Dyskrasie: nach Hippokrates falsche Zusammensetzung der Körpersäfte) bezeichnet.
Der Verdacht auf eine monoklonale Gammopathie ergibt sich aus einer auffälligen Serumelektrophorese mit der typischen Spitze (sog. M-Gradient), der definitive Nachweis erfordert eine Immunelektrophorese oder eine Immunfixation. Das pathologische Immunglobulin lässt sich teilweise auch im Urin nachweisen, in manchen Fällen findet sich kein komplettes Immunglobulinmolekül, sondern nur freie κ- oder λ-Leichtketten. Man spricht von einer Bence-Jones-Proteinurie.
Die häufigsten monoklonalen Plasmazellerkrankungen sind:
- Monoklonale Gammopathie unklarer Signifikanz (MGUS)
- Schwelendes Multiples Myelom (SMM, smoldering multiple myeloma)
- Multiples Myelom
- Morbus Waldenström (auch: Waldenströms Makroglobulinämie).
- Leichtketten (AL-) Amyloidose
- Leichtketten-Ablagerungs-Krankheit (Light Chain Deposition Disease)
MGUS und SMM sind Störungen, bei denen weder Symptome noch Endorganschäden nachweisbar sind. Beim multiplen Myelom sind Endorganschäden nachweisbar, meist Blutarmut (Anämie), Erhöhung des Calciumspiegels (Hypercalcämie), Störungen der Nierenfunktion und umschriebene Auflösungen des Knochengewebes (Osteolysen). Die AL-Amyloidose kann alle Organsysteme befallen, am häufigsten betroffen sind Herz (restriktive Kardiomyopathie), Niere (Nephrotisches Syndrom, Niereninsuffizienz), Leber, Verdauungstrakt und Peripheres Nervensystem. Die Makroglobulinämie Waldenström geht einher mit einer vermehrten Bildung von monoklonalem Immunglobulin M und kann zu Hyperviskositätssyndrom, Anämie, Schwellung von Lymphknoten (Lymphadenopathie) und Vergrößerung von Leber und Milz (Hepatosplenomegalie) führen. Die Leichtketten-Ablagerungs-Krankheit befällt in erster Linie die Nieren und führt innerhalb weniger Jahre zum terminalen dialysepflichtigen Nierenversagen.
Diagnostische Kriterien und klinischer Verlauf monoklonaler Plasmazell-Erkrankungen[1] Erkrankung Diagnostische Kriterien Klinik und Verlauf Monoklonale Gammopathie unklarer Signifikanz (MGUS) - Monoklonales Protein im Serum <3g/dl
- Plasmazellen im Knochenmark <10%
- Keine Endorganschäden, die auf die Plasmazellerkrankung zurückgeführt werden können
- Keine Symptome
- 1% pro Jahr entwickeln sich zu multiplem Myelom oder verwandten Malignomen
Schwelendes Multiples Myelom (Smoldering Multiple Myeloma, SMM) - Monoklonales Protein im Serum ≥3g/dl
- und/oder Plasmazellen im Knochenmark ≥10%
- Keine Endorganschäden, die auf die Plasmazellerkrankung zurückgeführt werden können
- Keine Symptome
- in den ersten 5 Jahren entwickeln sich 10% pro Jahr zu multiplem Myelom,
- in den nächsten 5 Jahren entwickeln sich 3% pro Jahr zu multiplem Myelom,
- nach 10 Jahren entwickeln sich 1% pro Jahr zu multiplem Myelom.[2]
Multiples Myelom - Plasmazellen im Knochenmark ≥10%
- Monoklonales Protein in Serum und/oder Urin (Ausnahme: nicht sezernierendes Myelom)
- Endorganschäden: umschriebenen Konchenauflösungen (Osteolysen), Blutarmut (Anämie), erhöhter Calciumspiegel (Hypercalcämie), Nierenversagen
- Nachweis von Endorganschäden ist Voraussetzung zur Diagnose
- Mittlere Überlebenszeit ca. 4 Jahre
Morbus Waldenström - Monoklonales Immunglobulin M im Serum
- Typische Plasmazellen im Knochenmark ≥10% (lymphoplasmazelluläre Infiltration)
- Klinische Befunde: Erhöhte Viskosität des Blutplasmas (Hyperviskositätssyndrom), Blutarmut (Anämie), Lymphknotenschwellung (Lymphadenopathie), Vergrößerung von Leber und Milz (Hepatosplenomegalie)
- Mittlere Überlebenszeit ca. 5-6 Jahre
AL-Amyloidose - Systemerkrankung mit Beteiligung von Niere, Leber, Herz, Magen-Darm-Trakt, peripherem Nervensystem
- Nachweis von Amyloid in Geweben durch den Farbstoff Kongorot
- Nachweis von freien Leichtketten, meist Isotyp Lambda (λ) in den Amyloid-Ablagerungen
- Nachweis einer monoklonalen Plasmazell-Erkrankung
- Jedes Organ kann befallen sein, meist sind Herz, Nieren, periphere Nerven, Magen-Darm-Trakt und Leber betroffen
- Mittlere Überlebenszeit ca. 2 Jahre
Leichtketten-Ablagerungs-Krankheit (Light Chain Deposition Disease) - Niere ist immer betroffen, in 35% Beteiligung weiterer Organe, meist Leber und Herz, seltener peripheres Nervensystem und weitere Organe
- Kein Nachweis von Amyloid in Geweben durch den Farbstoff Kongorot
- Nachweis von freien Leichtketten, meist Isotyp Kappa (κ) in den Basalmembranen von Nierenkanälchen (Tubuli) und Nierenkörperchen (Glomeruli)
- Nachweis einer monoklonalen Plasmazell-Erkrankung
- In 37-65% der Fälle Nachweis eines multiplen Myeloms
- Mittlere Zeit bis zum Eintreten des terminalen (dialysepflichtigen) Nierenversagen 2,7 Jahre
- Mittlere Überlebenszeit ca. 2-5 Jahre
Einzelnachweise
- ↑ N. Leung, S. V. Rajkumar, : „Renal Manifestations of Plasma Cell Disorders.“ American Journal of Kidney Diseases 2007; 50: S. 155-165 Artikel
- ↑ Kyle, Robert A.: „Clinical Course and Prognosis of Smoldering (Asymptomatic) Multiple Myeloma.“ N Engl J Med 2007; 356: S. 2582-2590 Abstract

Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.