- Reaktionskinetik
-
Die Kinetik ist ein Teilbereich der physikalischen Chemie. Sie beschäftigt sich mit dem zeitlichen Ablauf chemischer Reaktionen (Reaktionskinetik) oder physikalisch-chemischer Vorgänge (z. B. Diffusion, Stoffabscheidung an Oberflächen). Die Kinetik unterteilt sich in zwei Teilbereiche, die Mikrokinetik und die Makrokinetik. Während die Mikrokinetik sich lediglich mit dem zeitlichen Ablauf einer Reaktion beschäftigt, wird in der Makrokinetik der Einfluss von makroskopischem Wärme- und Stofftransport einbezogen. In diesem Artikel soll nur auf die Mikrokinetik eingegangen werden.
Inhaltsverzeichnis
Die Reaktionsgeschwindigkeit
Die grundlegende Größe, mit der in der Kinetik gearbeitet wird, ist die Reaktionsgeschwindigkeit. Sie gibt an, wieviele Teilchen pro Zeit in einer chemischen Reaktion umgesetzt werden. Diese Geschwindigkeit hängt dabei von vielen Faktoren ab. Je nach zugrunde liegendem Modell gibt es unterschiedliche Möglichkeiten, die Reaktionsgeschwindigkeit zu betrachten.
Ein wichtiger Faktor, der zu berücksichtigen ist, ist die Konzentration der vorliegenden Stoffe. Je mehr Teilchen in einem Volumen vorliegen, desto mehr Kollisionen werden pro Zeiteinheit vorkommen. Da eine Reaktion aber nur stattfinden kann, wenn zwei Teilchen miteinander kollidieren, steigt die Reaktionsgeschwindigkeit mit der Konzentration der Edukte.
Wenn eine Reaktion folgenden Typs vorliegt
so gilt für die Hinreaktion das Geschwindigkeitsgesetz
wobei vR die Reaktionsgeschwindigkeit, ( − ΔcA) die Abnahme der Konzentration des Stoffes A und Δt die verstrichene Zeit ist. Diese Reaktionsgeschwindigkeit ist die Durchschnittsgeschwindigkeit der Reaktion, da einzelne Moleküle unterschiedliche lange Zeitintervalle benötigen, bevor sie in ein Reaktionsereignis eintreten.
Da die Abnahme der Edukte der Zunahme der Produkte entsprechen muss, gilt außerdem
Dieses stark vereinfachte Modell bedarf noch einiger Verfeinerungen bezüglich:
- der Aktivität, also die effektive Konzentration
- der Menge der Edukte im Verhältnis zu der Menge der Produkte und gegebenenfalls des Lösungsmittels
- der Temperatur
- der Stoßenergie
- der Anwesenheit von Katalysatoren
- der Reaktionen mit auftretenden Gasen vom Partialdruck
- der Ausrichtung großer Reaktionspartner (Enzyme, Katalysatoroberfläche) beim Zusammenstoß
- des Zerteilungsgrades
Die Reaktionsordnung
Für jede Reaktion lässt sich eine Reaktionsgleichung formulieren, die beschreibt, wieviele Edukt-Teilchen miteinander reagieren, um eine bestimmte Anzahl Produkt-Teilchen zu bilden.
Läge etwa folgende Reaktionsgleichung vor
würde dies bedeuten, dass zwei Teilchen A mit einem Teilchen B, einem Teilchen C und einem Teilchen D kollidieren müssten, um das Produkt E zu bilden.
Jedoch ist die Wahrscheinlichkeit, dass fünf Teilchen gleichzeitig und zudem mit ausreichender Energie kollidieren, äußerst gering.
Viel wahrscheinlicher ist, dass zunächst zwei oder drei Teilchen zusammen treffen, ein Zwischenprodukt bilden, dieses Zwischenprodukt kollidiert dann mit weiteren Teilchen gegebenenfalls unter Bildung weiterer Zwischenprodukte und zuletzt bildet sich das Produkt E, hier ein Beispiel:
Die Zerlegung der Gesamtreaktion in einzelne Schritte, in Elementarreaktionen, und deren Untersuchung zeigt, wie die Reaktion genau abläuft.
Experimentell oder auf der Grundlage modellhafter Annahmen kann ermittelt werden, wie die Reaktionsgeschwindigkeiten der Elementarreaktionen von den jeweiligen Konzentrationen der Komponenten A, B, C und D abhängen.
Die Abhängigkeit der Reaktionsgeschwindigkeit vom Exponenten mit dem die Konzentration eines bestimmten Reaktanden in das Geschwindigkeitsgesetz eingeht, wird als Reaktionsordnung in Bezug auf diesen Reaktanden bezeichnet.
Die Gesamtordnung einer Reaktion ist die Summe der Reaktionsordnungen aller an ihr beteiligten Reaktanden.
BeispielAngenommen, die Reaktionsgeschwindigkeit der ersten obigen Elementarreaktion hängt quadratisch von der Konzentration der Komponente A ab, ist diese Reaktion zweiter Ordnung in Bezug auf die Komponente A. Da keine sonstigen Teilchen an dieser Reaktion beteiligt sind, ist die Gesamtordnung der Reaktion ebenso zwei.
Angenommen, die Reaktionsgeschwindigkeit der zweiten obigen Elementarreaktion hängt linear von der Konzentration von A2 ab, linear von der Konzentration von B und gar nicht von der Konzentration von C ab. Dann ist die Reaktion erster Ordnung in Bezug auf A2, erster Ordnung in Bezug auf B und 0. Ordnung in Bezug auf C. Die Gesamtordnung ist auch hier zwei.
Angenommen, die Reaktionsgeschwindigkeit der dritten Elementarreaktion hängt linear von der Konzentration von A2BC, aber nicht von der Konzentration von D ab, so liegt eine Reaktion erster Ordnung in Bezug auf A2BC und eine nullter Ordnung in Bezug auf D vor. Die Gesamtordnung ist eins.
Die vier häufigsten, weil wahrscheinlichsten, Reaktionsordnungen sind:Reaktionen nullter Ordnung
Derartige Reaktionen sind unabhängig von der Konzentration der Reaktanden, hier ist die Reaktionsgeschwindigkeit also konstant. Dies kann z. B. der Fall sein, wenn es sich um Lichtabhängige Reaktionen handelt (dann ist der Faktor k von der Lichtintensität abhängig) aber auch nur, wenn das Photon als Teilchen und nicht als Welle betrachtet wird (was problematisch ist, wenn das Photon in der Reaktion verbraucht wird).
wobei
- v – Reaktionsgeschwindigkeit
- CA(t) – Konzentration des Stoffes A zum Zeitpunkt t
- t – Zeit
- k – Geschwindigkeitskoeffizient
Beispiele sind photochemische Reaktionen oder katalytische Reaktionen.
Bei Reaktionen pseudo-nullter Ordnung ist die Reaktion zwar abhängig von der Konzentration der Reaktanden, jedoch muss einer dabei erst aus einer mit ihm in einem dynamischen Gleichgewicht stehenden anderen Form, die in so großem Überschuss vorliegt, gebildet werden, so dass die Konzentration als konstant angesehen werden kann. Beispiel hierfür ist die Iodierung von Aceton bei dem nur die Enolform reagiert, aber die Ketoform zu nahezu 100 % vorliegt.
Reaktionen erster Ordnung
Hier handelt es sich um katalytische oder radioaktive Zerfallsprozesse. Die Reaktionsgeschwindigkeit ist nur von der Konzentration des zerfallenden Stoffes abhängig.
integriert
mit- [A]t – Konzentration von A zur Zeit t
- [A]0 – Anfangskonzentration von A
Reaktionen zweiter Ordnung
In diesem Falle reagieren zwei Edukte zu einem oder mehreren Produkten (Edukte und Produkte werden gemeinsam als Reaktanden bezeichnet). Die Reaktionsgeschwindigkeit ist abhängig von den Konzentrationen der Ausgangsstoffe.
oder bei nur einem Stoff
integriert
wobei
- [A] – Konzentration des Stoffes A
- [B] – Konzentration des Stoffes B
Die meisten bimolekularen Reaktionen in flüssigem oder festem Medium folgen dieser Kinetik.
Allerdings gibt es einen Sonderfall, bei dem einer der Reaktanden in einem sehr hohen Überschuss vorliegt, so dass die Konzentrationsänderung über die Zeit der Reaktion verschwindend gering ist. Das ist zum Beispiel der Fall, wenn Wasser sowohl Reaktionspartner als auch das Lösungsmittel darstellt (z. B. bei einer Esterhydrolyse). In diesem Fall folgt die Reaktionsgeschwindigkeit den Gesetzmäßigkeiten einer Reaktion erster Ordnung. Da es sich aber trotzdem um eine bimolekulare Reaktion handelt, spricht man von Reaktionen pseudoerster Ordnung (oder scheinbar erster Ordnung).
Ein weiterer Sonderfall ergibt sich, wenn experimentell eine Reaktion 0. Ordnung gemessen wird (obwohl eine Reaktion höherer Ordnung vorliegt). Dann spricht man von einer Reaktion von scheinbar nullter Ordnung. Dies kann insbesondere bei katalytischen Prozessen der Fall sein (wie z. B. der Enzymkatalyse oder der katalytischen Hydrierung von Ethen) wenn der limitierende Faktor die Anzahl „Plätze“ am Katalysator sind und nicht wie oft die Moleküle zusammenprallen (da dann die direkte Elementarreaktion so gut wie keine Rolle mehr spielt).
Reaktionen dritter Ordnung
In diesem Falle reagieren drei Reaktanden zu einem oder mehreren Produkten. Eine trimolekulare Reaktion ist sehr selten (rein statistisch ist die Wahrscheinlichkeit sehr gering, dass drei Teilchen zur gleichen Zeit am gleichen Ort sind) und in vielen Fällen wird eine konkurrierende (und dominante) bimolekulare Reaktion beobachtet. Ein Beispiel einer wirklich trimolekularen Reaktion ist die Atomrekombination. Die Reaktionsgeschwindigkeit ist von drei Stoffen abhängig:
- Für die Elementarreaktion
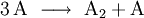 lautet das Geschwindigkeitsgesetz
lautet das Geschwindigkeitsgesetz
man beachte, dass der Faktor 1/2 von der stöchiometrischen Gleichung 2A = A2 kommt und nicht von der Elementarreaktion (währenddessen aber der Exponent 3 von der stöchiometrischen Gleichung herrührt).
- Integrierte Form
![\frac {1}{ {[A]_0}^{2} } - \frac {1}{ {[A]_t}^{2} } = -4k*t](/pictures/dewiki/98/bf6d74aef472a071d710b77b9744101d.png)
Ein Beispiel für diese Reaktion:
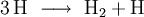
Temperaturabhängigkeit der Reaktionsgeschwindigkeit
RGT-Regel (Reaktionsgeschwindigkeit-Temperatur-Regel): Wird die Temperatur für eine chemische Reaktion um 10 K erhöht, dann erhöht sich die Reaktionsgeschwindigkeit um das 2- bis 4-fache (für eine Reaktion mit einer Aktivierungsenergie um die 50 kJ/mol!)
Mathematisch und physikalisch wird dies mit dem Ansatz von Arrhenius begründet. Nach diesem kann die Temperaturabhängigkeit der Reaktionsgeschwindigkeit mit einer Exponentialfunktion beschrieben werden. Wie oben schon beschrieben lautet die Bestimmungsgleichung für eine Reaktion zweiter Ordnung:
Die Temperaturabhängigkeit ist in dem so genannten Geschwindigkeitskoeffizienten enthalten, denn es gilt hier die Arrhenius-Gleichung. Der Geschwindigkeitskoeffizient k ist nur dann eine Konstante, solange die Temperatur nicht verändert wird und kann dann auch als Geschwindigkeitskonstante bezeichnet werden.
Überblick über die wichtigsten Formeln der Reaktionskinetik Reaktionstyp Berechnung der Momentan-
geschwindigkeit vBerechnung der
Halbwertszeit0. Ordnung 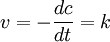
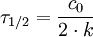
1. Ordnung 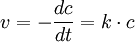
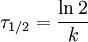
2. Ordnung 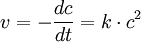
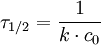
Kinetische Untersuchungen
Zur Untersuchung der Kinetik chemischer Reaktionen steht eine Vielzahl von Verfahren zur Verfügung.
Diese ermöglichen die Bestimmung der
- Ordnung der Reaktion
- Elementarreaktionen
- Reaktionsgeschwindigkeitskonstanten
- Temperaturabhängigkeit und Aktivierungsenergie
- des Arrhenius-Faktors
Messverfahren
Aus der Vielzahl der Messverfahren sollte natürlich immer dasjenige ausgewählt werden, das die zuverlässigsten Ergebnisse liefert und am einfachsten durchzuführen ist. Dazu muss man die physikalischen Eigenschaften der Reaktionskomponenten berücksichtigen, sowie die Reaktionsbedingungen und vor allem die Geschwindigkeit/Halbwertszeit der zu untersuchenden Reaktion.
Generell gilt:
- Bei Kenntnis der Reaktionsgleichung reicht es, den Umsatz einer Komponente zu beobachten.
- Die Messgröße muss quantitativ proportional zur Konzentration/Stoffmenge der beobachteten Komponente(n) sein.
- Bei den meisten Verfahren sollte für die maximale Durchmischung der Reaktionspartner gesorgt werden.
- Die Analysenzeit sollte wesentlich kürzer sein als die Reaktionszeit.
- Das Messverfahren sollte den Reaktionsablauf möglichst nicht beeinflussen, bzw. der Einfluss des Messverfahrens sollte klein gehalten werden und bei der Analyse berücksichtigt werden.
Man unterscheidet:
- kontinuierliche Messverfahren, bei denen die Konzentration/Stoffmenge der Komponente(n) durchgehend am Reaktionsgemisch selbst beobachtet wird, z. B. Spektralphotometrie.
- diskontinuierliche Messverfahren, bei denen die Messung entweder in Intervallen am Reaktionsgemisch durchgeführt wird oder an genommenen Proben des Reaktionsgemisches zu definierten Zeitpunkten.
Diskontinuierliche Messverfahren sind nur bei langsamen Reaktionen (Minuten- bis Stundenbereich) zu empfehlen. Außerdem muss hier darauf geachtet werden, dass die Messung unmittelbar nach der Probenentnahme erfolgt, damit die Konzentration/Stoffmenge der weiterhin reagierenden Substanzen nicht verfälscht wird.
Um diesen Effekt zu senken, kann man die Probe stark kühlen (Absenkung der Reaktionsgeschwindigkeit), oder eine der reagierenden Komponenten aus der Probe entfernen, z. B. durch Fällung.
Bei schnellen Reaktionen (Minuten- bis Sekundenbereich) ist ein kontinuierliches Messverfahren notwendig. Oft werden hier spezielle Strömungsapparaturen verwendet.
Bei extrem schnellen Reaktionen (Millisekundenbereich) werden Spezialverfahren angewendet, z. B. Relaxationsverfahren oder die Blitzlichtphotolyse.
Messgrößen
Zur Messung des Umsatzes einer Komponente sollte eine Messgröße verwendet werden, die möglichst eindeutig und proportional mit der Konzentration/Stoffmenge einer Komponente in Verbindung gebracht werden kann. Dazu ist eine Kalibrierung notwendig.
Weiterhin ist wichtig, unter welchen Bedingungen die Reaktion durchgeführt wird, also isobar bei konstantem Druck, isotherm bei konstanter Temperatur oder isochor bei konstantem Volumen.
Die am häufigsten genutzten Messgrößen sind:
- Dielektrizitätskonstante
- Leitfähigkeit bei ionischen Spezies
- Brechzahl
- optische Aktivität bei optisch aktiven Spezies
- Lichtabsorption bei lichtabsorbierenden Spezies (häufigstes Messverfahren)
- Fluoreszenz
All diese Messgrößen können sowohl kontinuierlich als auch diskontinuierlich beobachtet werden und eignen sich damit für schnelle und langsame Reaktionen.
Bei langsamen Reaktionen kann man außerdem messen:
- Volumen bei isobaren Bedingungen bei Reaktionen mit Gasentwicklung/verbrauch
- Druck bei isochoren Bedingungen, bei Gasen
- Wärmetönung (selten)
Da alle Messgrößen temperaturabhängig sind, sollte isotherm gearbeitet werden, d. h. die Kalibrierung sollte bei einer definierten Temperatur vorgenommen werden, die für die Messung beibehalten werden muss.
Mischverfahren
Bei langsamen bis schnellen Reaktionen werden hauptsächlich Mischverfahren eingesetzt. Hierbei werden definierte Stoffmengen der Reaktanden möglichst augenblicklich miteinander vermischt unter Verwendung einfacher Rührer, Strömungsrohre oder hochpräziser Mischkammern.
Die Konzentrationsänderungen werden kontinuierlich oder diskontinuierlich beobachtet.
Bekannt sind dann:
- die Ausgangskonzentration(en) [A]0
- unterschiedliche Konzentrationen [A]t zu den Zeitpunkten t
- die Reaktionstemperatur T
Im Folgenden sollten die Prinzipien der kinetischen Untersuchungen mit Hilfe der Mischverfahren vereinfacht dargestellt werden.Bestimmung der Reaktionsordnung
Bei der Methode der variierten Ausgangskonzentrationen werden die Anfangskonzentrationen der verschiedenen Reaktanden in mehreren Reaktionsansätzen variiert bei sonst gleich bleibenden Reaktionsbedingungen.
Pro Ansatz sollte am besten die Anfangskonzentration nur eines Reaktanden variiert werden, damit der Effekt eindeutig zugeordnet werden kann. Weiterhin müssen die Mischvolumina genau berechnet werden.
Bei allen Ansätzen wird nach einer definierten Reaktionszeit t der Umsatz eines Eduktes oder Produktes [A]t bestimmt und daraus die Reaktionsgeschwindigkeit berechnet:
Nun ergeben sich verschiedene Möglichkeiten:
- v verdoppelt sich, wenn die Anfangskonzentration eines Reaktanden verdoppelt wird: Abhängigkeit erster Ordnung von der Konzentration dieses Reaktanden.
- v vervierfacht sich, wenn die Anfangskonzentration eines Reaktanden verdoppelt wird: Abhängigkeit zweiter Ordnung von der Konzentration dieses Reaktanden.
- v bleibt gleich unabhängig von der Anfangskonzentration eines Reaktanden: Abhängigkeit nullter Ordnung von der Konzentration dieses Reaktanden.
usw.
Die Gesamtordnung ergibt sich aus der Summe der Einzelordnungen.
Bestimmung des Reaktionsgeschwindigkeitskoeffizienten
Es wird eine definierte Ausgangskonzentration [A]0 des Reaktanden A gewählt und dessen Konzentration [A]t wird mehrmals oder kontinuierlich bei der Reaktion beobachtet. Die Reaktionsordnung bzgl. A muss bekannt sein.
Zu verschiedenen Zeitpunkten t1, t2, t3 usw. erhält man unterschiedliche Konzentrationen [A]1, [A]2, [A]3 usw.
Diese Wertepaare und die Ausgangskonzentration kann man in das umgeformte Zeitgesetz der entsprechenden Reaktionsordnung einsetzen und graphisch auftragen (Arrhenius-Plot):
- 0. Ordnung:
- 1. Ordnung:
- 2. Ordnung:
In allen Fällen erhält man k aus der Steigung der entstehenden Graphen. Je mehr Messpunkte man hat, desto genauer wird das Ergebnis.Bei Reaktionen 0. Ordnung gibt es einen linearen Zusammenhang zwischen [A] und der Zeit t, bei Reaktionen 1. Ordnung zwischen ln [A] und der Zeit t, bei Reaktionen 2. Ordnung zwischen 1/[A] und der Zeit t. Indem man experimentell bestimmt, welche Funktion linear mit der Zeit läuft (man setzt die erhaltenen Konzentrationswerte in die verschiedenen Gleichungen der linearen Beziehungen ein), kann man die Ordnung einer Reaktion ermitteln.
Bestimmung des Arrhenius-Faktors und der Aktivierungsenergie
Nach obigem Verfahren bestimmt man k1, k2, k3 usw. bei mehreren Temperaturen T1, T2, T3 usw.
Durch Umformen der Arrhenius-Gleichung erhält man:
Bei Einsetzen der Wertepaare k(T) und graphischer Auftragung von ln(k) gegen
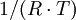 erhält man eine Gerade. Die Steigung entspricht dann dem Negativen der Aktivierungsenergie EA, der Y-Achsenabschnitt dem Logarithmus des Arrhenius-Faktors ln(A).
erhält man eine Gerade. Die Steigung entspricht dann dem Negativen der Aktivierungsenergie EA, der Y-Achsenabschnitt dem Logarithmus des Arrhenius-Faktors ln(A).Relaxationskinetik
Reaktionen mit Halbwertszeiten im Millisekundenbereich oder weniger lassen sich nur schwer beobachten; auf Mischverfahren wird hier deswegen ganz verzichtet.
Bei Relaxationsverfahren werden Reaktanden miteinander vermischt. Die Reaktion lässt man ablaufen, bis sich ein Reaktionsgleichgewicht eingestellt hat.
Dann wird das Gleichgewicht sprungartig gestört und man beobachtet, wie schnell sich das Gleichgewicht wieder einstellt, die Relaxation.
Nach der neuen Gleichgewichtseinstellung werden die Konzentrationen wieder bestimmt und die neue Gleichgewichtskonstante wird berechnet.
Relaxation und Relaxationszeit
Die Relaxation, also die Herstellung eines neuen Gleichgewichtes, läuft nach einer Exponentialfunktion ab, sofern die Abweichung von dem Gleichgewicht gering ist, der „Sprung“ also nicht zu groß war.
mit
- xt – Konzentrationsabweichung einer Komponente von der neuen Gleichgewichtskonzentration nach dem „Sprung“ zur Zeit t
- x0 – Konzentrationsabweichung einer Komponente von der neuen Gleichgewichtskonzentration zur Zeit des „Sprunges“
- τ – '
Die Relaxationszeit ist die Zeit bis die Abweichung vom Gleichgewicht xt auf x0 / e abgeklungen ist.Je nach Anzahl der Reaktanden und Reaktionsordnung, sowie bei Kenntnis der Gleichgewichtskonstanten des neu eingestellten Gleichgewichtes kann man aus τ die Geschwindigkeitskoeffizienten der Hin- und Rückreaktionen extrem schneller Prozesse berechnen.
Relaxationsverfahren
Um ein chemisches Gleichgewicht zu stören, bieten sich viele Variablen an. Wichtig jedoch ist, dass der „Sprung“ augenblicklich erfolgt und nicht zu groß ist.
Bewährt haben sich:
- Temperatursprungverfahren: schnelle Temperaturerhöhung durch Kondensatorentladung in einen Elektrolyten
- Drucksprungverfahren: schnelle Druckverringerung durch Bersten des Reaktionsraumes in einen größeren Reaktionsraum
- Feldsprungverfahren: schnelle Verringerung des elektrischen Feldes durch Abschalten einer Spannungsquelle in einem Elektrolyten
Blitzlichtphotolyse
Hierbei wird mit einer Blitzapparatur zunächst ein starker Lichtblitz erzeugt, der zur Photolyse einer chemischen Verbindung führt. Kurze Zeit später erfolgt ein zweiter, schwächerer Blitz, der der spektrophotometrischen Messung eines Reaktanden dient.
Dieses Verfahren eignet sich besonders für radikalische Prozesse.
Siehe auch
Literatur
Allgemeine Lehrbücher
Kinetik
- J. I. Steinfeld, J. S. Francisco, W. L. Hase, Chemical Kinetics and Dynamics, 2. Aufl., Prentice Hall, 1998, ISBN 978-0-13-737123-5.
- M. J. Pilling, P. W. Seakins, Reaction Kinetics, Oxford University Press, 1995, ISBN 978-0-19-855527-8.
Elektrochemische Kinetik
- W. Forker, Elektrochemische Kinetik, Akademie Verlag, Berlin, 1966.
- W. Vielstich, W. Schmickler, Elektrochemie II: Elektrochemische Kinetik, Dr. Dietrich Steinkopff Verlag, Darmstadt, 1976.
- K. Vetter, Elektrochemische Kinetik, Springer Verlag, Berlin, 1961.
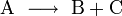
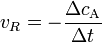
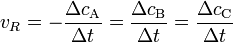
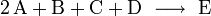
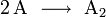
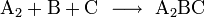
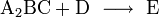
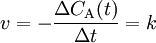
![v= -\frac {d[\mathrm{A}]}{dt}= k \cdot [\mathrm{A}]](/pictures/dewiki/51/3eae6de39c4daaaf476d31bb6132f099.png)
![\mathrm{[A]}_{t} = \mathrm{[A]}_{0} \cdot e^{-k\cdot t}](/pictures/dewiki/50/27a20f08a23e0eddb4da331464a5f4ba.png)
![v= -\frac {d\mathrm{[A]}}{dt}= -\frac {d\mathrm{[B]}}{dt}=k \cdot \mathrm{[A]}\cdot\mathrm{[B]}](/pictures/dewiki/48/096f5367477473a4c354e031d4780b63.png)
![v= - \frac {d\mathrm{[A]}}{dt}=k\cdot\mathrm{[A]}^{2}](/pictures/dewiki/54/62650714f181051853beb3105a3fdf64.png)
![\mathrm{[A]}_{t} = \frac {1} {k\cdot t + \frac {1} {\mathrm{[A]}_{0}}}](/pictures/dewiki/100/d8b7e4fbd5e1d05cce6a1b2df3912fbd.png)
![v= -\frac {1}{2} \frac {d\mathrm{[A]}}{dt}= k \cdot \mathrm{[A]}^{3}](/pictures/dewiki/56/82717cfbea56b224d90252e11d00a7af.png)
![v=k\cdot\mathrm{[A]}\cdot\mathrm{[B]}](/pictures/dewiki/97/a4404cfced0c39b7b0b347a8070fb21c.png)
![v= \frac {d \mathrm{[A]}} {dt} = \frac {\mathrm{[A]}_{t} - \mathrm{[A]}_{0}} {t}](/pictures/dewiki/100/dcc60b60e7f5071eb8489a2be83b82a5.png)
![\mathrm{[A]}_{0} - \mathrm{[A]}_{t} = k \cdot t](/pictures/dewiki/49/14c5ba2ca62d2321884db7f5aed5fba0.png)
![\ln{ \frac {\mathrm{[A]}_{0}} {\mathrm{[A]}_{t}}} = k \cdot t](/pictures/dewiki/48/05d237e809db2fd1cc70e678b4b1116f.png)
![\frac {1} {\mathrm{[A]}_{t}} - \frac {1} {\mathrm{[A]}_{0}} = k \cdot t](/pictures/dewiki/48/09c567b07c7300d18c7777508d3c85a5.png)
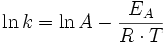
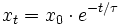
Wikimedia Foundation.