- Retinitis pigmentosa
-
Klassifikation nach ICD-10 H35.5 Hereditäre Netzhautdystrophie ICD-10 online (WHO-Version 2006) Die Bezeichnung Retinopathia pigmentosa oder Retinitis pigmentosa (RP) beschreibt eine durch Vererbung oder spontane Mutation entstehende Netzhautdegeneration, bei der die Photorezeptoren zerstört werden. Man spricht von Pseudo-Retinitis pigmentosa (oder Phänokopie), wenn nicht erbliche Erkrankungen Symptome der RP zeigen, z. B. toxisch bedingt (z. B. durch Phenothiazine, Chloroquin).
Der Name Retinitis pigmentosa wurde von dem Holländer Franz Donders im Jahr 1855 geprägt [1]. Da es sich bei RP nicht primär um eine Entzündung handelt, wurde die Krankheit in Retinopathia pigmentosa umbenannt. Die ursprüngliche Benennung wird allerdings immer noch synonym und sogar häufiger verwendet als die neue Bezeichnung.
Inhaltsverzeichnis
Epidemiologie
Weltweit sind etwa drei Millionen Menschen – in Deutschland etwa 30.000 bis 40.000 – von einer der verschiedenen Formen der RP betroffen. Bedingt durch den schleppenden Verlauf der Erkrankung dürfte die Dunkelziffer noch höher sein. Die Prävalenz liegt somit bei 1 Fall pro 3000 bis 7000 Einwohnern. [2]
Die Erkrankung tritt meistens im Jugendalter oder in den mittleren Lebensjahren mit den ersten Merkmalen (Nachtblindheit) ein; die Sehkraft lässt allmählich nach. Der gesamte Prozess zunehmender Sehbehinderung verläuft schleichend und erstreckt sich beim Betroffenen meistens über Jahrzehnte hinweg. Diese Entwicklung ist so auch mit einer starken psychischen Belastung verbunden. Bei ca. der Hälfte aller RP-Patienten entwickelt sich im Erwachsenenalter eine Linsentrübung (grauer Star). [3]
Symptome und Verlauf
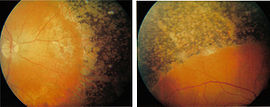 Augenhintergrund eines Patienten mit Retinopathia pigmentosa (in diesem Fall Refsum-Syndrom)
Augenhintergrund eines Patienten mit Retinopathia pigmentosa (in diesem Fall Refsum-Syndrom)Bei RP treten in der Regel folgende Symptome auf:
- Nachtblindheit
- schlechte Anpassung der Augen auf sich ändernde Lichtbedingungen
- Blendempfindlichkeit
- Einschränkung des Gesichtsfeldes besonders in der Peripherie, das zentrale Gesichtsfeld bleibt erhalten. ("Röhrenblick" oder "Tunnelblick" oder "Schrotflinten-Gesichtsfeld")
- Störung des Kontrastsehens
- Störung des Farbsehens
Im Verlauf kommt es typischerweise erst zu Nachtblindheit, Visusabfall und dann zu einer langsamen Einschränkung des Gesichtsfeldes bis hin zu einem sich immer mehr verengenden „Tunnelblick“, was in einem späteren Stadium in der Regel zur Blindheit führt. Durch den Tunnelblick können sich die Patienten kaum mehr im Raum orientieren und nicht mehr alleine gehen. Der zeitliche Verlauf dieser Abfolge variiert dabei, abhängig vom jeweiligen RP-auslösenden Gendefekt. [4]
Das Absterben der Photorezeptoren, zuerst der Stäbchen, vollzieht sich in der Regel von der Peripherie (dem Rand des Gesichtsfeldes) zur Makula (dem Zentrum des Gesichtsfeldes) hin. Donders beschrieb besonders die Knochenkörperchen-ähnlichen Pigmenteinlagerungen und Gefäßverengungen im Auge, welche der RP den Namen gaben. Diese Veränderungen treten aber in der Regel sekundär zur Degeneration der Photorezeptoren auf.
Genetik
Über 45 Gene wurden inzwischen identifiziert, deren Defekt RP auslösen kann. Dennoch konnten bislang nur etwas über die Hälfte aller verursachenden Erbfaktoren identifiziert werden. Die meisten bislang identifizierten Gene folgenden einem monogentischem Erbgang, d. h. der Defekt von nur einem Gen verursacht bereits die Erkrankung und nicht mehrerer Genen gleichzeitig. Die Erkrankung wird sowohl durch autosomal dominante, autosomal rezessive und auch gonosomale Erbfaktoren (im wesentlichen das X-Chromosom) ausgelöst. [2][3]
Assoziierte R. pigmentosa oder Syndrome
Ungefähr 25% der betroffenen Patienten leiden an einer assoziierten RP. Bei der assoziierten RP weisen neben dem Auge auch andere Organe des Körpers Krankheitssymptome auf, das heißt, es liegt ein Syndrom vor [2]. Einige solcher mit RP oftmals zusammen auftretenden Symptome sind Hörstörungen, Lähmungen und Gehstörungen, Herzrhythmusstörungen, Muskelschwäche, geistige Entwicklungsstörungen, u. a.. Auch hier sind Gendefekte die Ursache. Die bekanntesten Syndrome sind:
- das Usher-Syndrom
- das Bardet-Biedl-Syndrom
- das Refsum-Syndrom
- das NBIA-Syndrom
- das Alport-Syndrom
Diagnose und Behandlung
Bereits in früher Kindheit kann eine RP über ein Elektroretinogramm diagnostiziert werden. Weitere Diagnosemöglichkeiten bieten Sehtests zur Nachtblindheit beim Augenarzt. Bei Syndromen geben die weiteren Symptome Hinweise auf die genaue Erkrankung (wie Hörstörungen oder Blutwerte). Die Ermittlung des genauen Gendefekts ermöglicht erst eine DNA-Analyse. Noch in der Entwicklung befinden sich DNA-Chip und Protein-Chip, welche eine schnellere Diagnose ermöglichen sollen. Dies kann auch zur genetischen Familienberatung beitragen.
Es gibt derzeit keine Behandlung, die das Fortschreiten von RP verhindern oder die Krankheit heilen kann. Allerdings existieren Studien, wonach die Einnahme von Vitamin A den Verlauf verlangsamen soll. [5] Eine Ausnahme bilden Sonderformen wie z. B. das Refsum-Syndrom, ein Stoffwechseldefekt, bei dem eine phytansäurearme Spezialdiät die RP zum Stillstand bringen kann.
Noch in der Erforschung befinden sich gentherapeutische Ansätze, bei denen defekte Gene in der Retina ersetzt werden könnten, oder Stammzelltherapien, bei denen die degenerierte Retina repariert werden soll. [5][6]. In der Entwicklung sind auch sogenannte Retina-Implantate, bei denen Mikrosystemtechnik als Prothese die Funktionen der defekten Retina ersetzen soll [7]. Aktuell sollen so mit einem subretinalen Implantat mit einer möglichen Auflösung von 1500 Dioden wieder erste Seherfolge möglich sein[8].
Wie auch bei anderen Erkrankungen ohne wissenschaftlich anerkannte Heilmethode nehmen manche Patienten alternativmedizinische Therapien in Anspruch. Trotz vieler Studien in den letzten Jahrzehnten ist auch auf diesem Gebiet ein Wirkungsnachweis bisher nicht gelungen. Durch Maßnahmen der Rehabilitation kann eine weitgehend selbständige Lebensführung erreicht werden.
Weblinks
- Themenausgabe zur Retinitis Pigmentosa im Online-Magazin sciencegarden
Referenzen
- ↑ F.C. Donders: Beiträge zur pathologischen Anatomie des Auges. Arch für Ophthalmol 1855;1: 106-118
- ↑ a b c S.P. Daiger et al.: Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. 2007 Feb;125(2):151-8. PMID 17296890
- ↑ a b D.T. Hartong et al.: Retinitis pigmentosa. Lancet. 2006 Nov 18; 368(9549): 1795-809. PMID 17113430
- ↑ M.A. Sandberg et al.: Disease Course in Patients with Autosomal Recessive Retinitis Pigmentosa due to the USH2A Gene. Invest Ophthalmol Vis Sci. 2008 Jul 18. PMID 18641288
- ↑ a b P. Goodwin: Hereditary retinal disease. Curr Opin Ophthalmol. 2008 May;19(3):255-62. PMID 18408503
- ↑ I. Mooney I, J. LaMotte: A review of the potential to restore vision with stem cells. Clin Exp Optom. 2008 Jan;91(1):78-84. PMID 18045253
- ↑ N. Alteheld et al.: Towards the bionic eye--the retina implant: surgical, opthalmological and histopathological perspectives. Acta Neurochir Suppl. 2007;97(Pt 2):487-93. PMID 17691339
- ↑ Pilotstudie erfolgreich: Chip ermöglicht Blinden erste Orientierung. BioPro 2007 [1]

Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.