- Retinoblastom
-
Klassifikation nach ICD-10 C69.2 Bösartige Neubildung: Retina ICD-10 online (WHO-Version 2011) 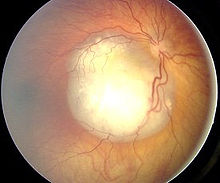 Fundusansicht mit Retinoblastom
Fundusansicht mit Retinoblastom
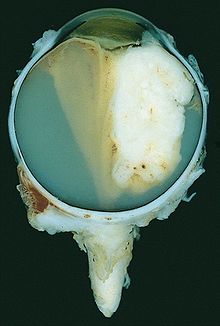 Großes exophytisch wachsendes Retinoblastom mit Verkalkungen und massiver Ablösung der Netzhaut
Großes exophytisch wachsendes Retinoblastom mit Verkalkungen und massiver Ablösung der NetzhautDas Retinoblastom ist ein bösartiger Tumor in der Netzhaut des Auges, für dessen Entstehung Mutationen in beiden Allelen des Retinoblastom-Gens (lokalisiert auf Chromosom 13, Bande q14) die Grundvoraussetzung sind.
Dieser Tumor geht von genetisch veränderten unreifen Netzhautzellen aus und führt unbehandelt zum Tode. Wird die Krankheit frühzeitig erkannt und therapiert, sind die Heilungschancen gut (ca. 95 % der Patienten werden geheilt). Da das Wachstum des Retinoblastom nur von unreifen Netzhautzellen ausgehen kann, kann dieser Tumor auch nur bis zum 5. Lebensjahr auftreten. Auf 20.000 Lebendgeburten kommt etwa ein Krankheitsfall, was ca. 60 Fälle pro Jahr in Deutschland entspricht. In den USA rechnet man pro Jahr mit etwa 350 Fällen, weltweit geht man von 5.000-8.000 Neuerkrankungen pro Jahr aus. Zu unterscheiden ist eine erbliche von einer nicht-erblichen Form. Bei Mädchen und Jungen tritt der Tumor mit gleicher Häufigkeit auf. Abgesehen vom Menschen ist bisher keine Tierart bekannt, bei der ein Retinoblastom natürlicherweise auftritt.
Inhaltsverzeichnis
Ursache
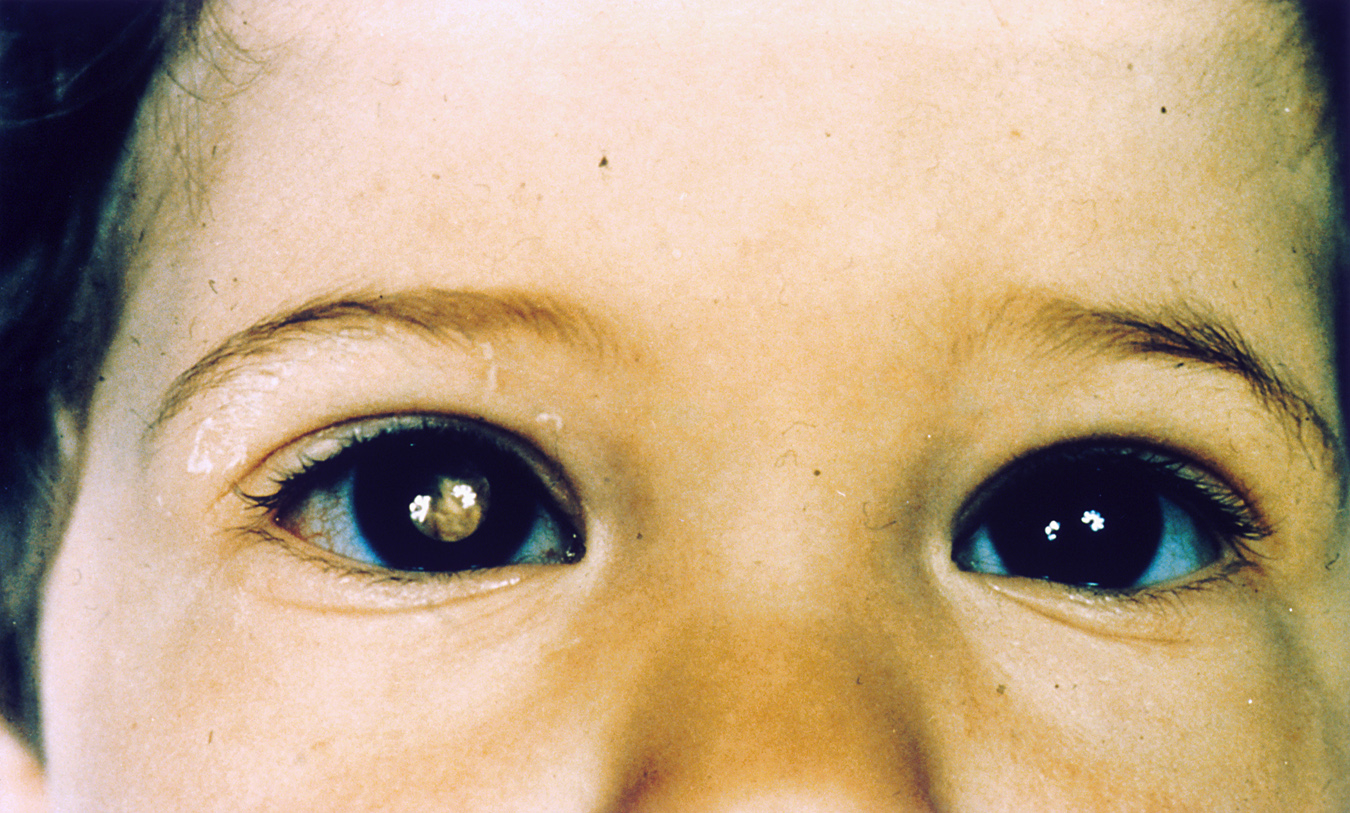
 Durch die Pupille des rechten Auges kann man einen gelblichen intraokulären Tumor erkennen
Durch die Pupille des rechten Auges kann man einen gelblichen intraokulären Tumor erkennen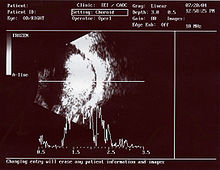 Ultraschallbild eines Retinoblastoms
Ultraschallbild eines RetinoblastomsEtwa 45 % der Patienten haben die erbliche Form des Retinoblastoms. Diese Patienten sind heterozygot für eine Mutation im Retinoblastom-Gen (erste Mutation). Diese Mutationen sind meist das Resultat von Neumutationen in der Keimbahn (Samenzelle bzw. Eizelle) eines Elternteils. Es liegt ein autosomal-dominanter Erbgang mit unvollständiger, aber hoher Penetranz vor. Dieser autosomal-dominante Erbgang ist insofern bemerkenswert, als er sich von sonstigen autosomal-dominanten Erbgängen deutlich unterscheidet. Zur Entstehung des Retinoblastoms müssen beide Allele für das Retinoblastomgen (RB1) mutiert sein, dies ist normalerweise eine Eigenschaft, die autosomal-rezessiv vererbte Krankheiten aufweisen. Im Falle des erblichen Retinoblastoms jedoch kommt es in nahezu 95 % der Fälle zu der zusätzlichen zweiten somatischen Mutation in Retinavorläuferzellen bei bestehender erster Mutation aller Körperzellen. Der mutationsbedingte Verlust des zweiten, normalen Allels in einer Vorläuferzelle der Retina löst die Entstehung des Tumors aus. Dadurch erscheint der Erbgang typisch autosomal-dominant mit nicht vollständiger, aber hoher Penetranz. Untersuchungen hierzu wurden von Alfred G. Knudson durchgeführt und 1971 veröffentlicht, nach ihm heißt diese Theorie zur Tumorentstehung „Zwei-Mutationen-Theorie nach Knudson“. Inzwischen geht man davon aus, dass ein dritter Faktor hinzukommen muss, damit es zur Tumorbildung kommt. Diskutiert werden in diesem Zusammenhang beispielsweise Veränderungen in der Chromosomenstruktur, wie beispielsweise Zugewinne auf dem langen Arm von Chromosom 1 oder auf dem kurzen Arm des Chromosom 6.
Die meisten Patienten mit erblichem Retinoblastom haben Tumoren in beiden Augen (beidseitiges Retinoblastom). Heterozygote Träger einer Mutation im Retinoblastom-Gen geben diese mit einem Risiko von 50 % an die Nachkommen weiter. Bei Kindern, die eine solche Mutation geerbt haben, besteht ein sehr hohes Risiko für die Entwicklung von Retinoblastomen.
Etwa 55 % der Patienten haben die nicht-erbliche Form des Retinoblastoms. Bei diesen Patienten treten beide für die Tumorentstehung erforderlichen Mutationen in Körperzellen auf (somatische Mutationen), und zwar müssen beide Mutationen in ein und derselben Zelle vorliegen. Da es statistisch gesehen recht unwahrscheinlich ist, dass diese zweifache Mutation bei einem Kind unabhängig voneinander in Zellen der Retina beider Seiten passiert, ist das sporadische Retinoblastom selten bilateral. Fast alle Patienten mit nicht-erblichem Retinoblastom haben nur ein betroffenes Auge (einseitiges Retinoblastom).
Da das Retinoblastom meist bei jungen Kindern unter fünf Jahren auftritt, wird das Retinoblastom auch als Kindlicher Augentumor bezeichnet. Die Erkrankung wird bei Kindern mit beidseitigem Retinoblastom überwiegend früher als bei Kindern mit einseitigem Retinoblastom festgestellt; das durchschnittliche Alter bei Diagnosestellung liegt bei der unilateralen Form bei 23 Monaten, bei der bilateralen Form bei 12 Monaten. Etwa 10 % der Retinoblastome werden bereits kurz nach der Geburt diagnostiziert, innerhalb des ersten Lebensjahres etwa 50 %, und bis zum 3. Lebensjahr ungefähr 90 %.
Sonderformen
Trilaterales Retinoblastom
 Trilaterales Retinoblastom, aufgenommen mittels Magnetresonanztomographie
Trilaterales Retinoblastom, aufgenommen mittels MagnetresonanztomographieTrilaterales Retinoblastom nennt man ein sehr selten vorkommendes erbliches Retinoblastom, das gemeinsam mit einem Hirntumor auftritt. Dabei handelt es sich um eine selbständige Geschwulst, nicht um eine Metastase. Die Histologie ähnelt der des Retinoblastoms; die Prognose ist für den Patienten relativ ungünstig.
Retinom (Retinozytom)
Retinom bezeichnet einen gutartigen (benignen) Netzhauttumor, der in ungefähr 2 % der Patienten mit einer Mutation des RB-Gens auftritt. Es handelt sich dabei vermutlich um einen Vorläufer des Retinoblastoms oder um die Reste eines abgeheilten Retinoblastoms nach spontaner Regression. Die Histologie ist ähnlich der des Retinoblastoms.
Symptomatik
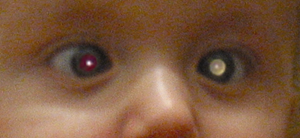
 Typische Leukokorie bei einem Kind mit Retinoblastom
Typische Leukokorie bei einem Kind mit Retinoblastom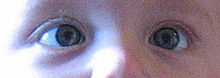 Schielstellung, verursacht durch ein Retinoblastom
Schielstellung, verursacht durch ein RetinoblastomSymptom Häufigkeit Leukokorie 56 % Strabismus 20 % schmerzhafte Rötung/Glaukom 7 % Sehverlust 5 % orbitale Cellulitis 3 % unilaterale Mydriasis 2 % Heterochromia iridis 1 % Hyphäma 1 % weiße Irisflecken 0,5 % Anorexie/Entwicklungsstörung 0,5 % Nicht selten fällt der Tumor durch sogenannte Leukokorie auf, dabei wird das in das Auge einfallende (Blitz-)Licht nicht wie beim gesunden Auge von der Netzhaut mit der darunterliegenden Aderhaut reflektiert (dies ergibt das typische „rote Auge“ auf Photographien), sondern vom Retinoblastomgewebe (hierdurch kommt es zu einer weißlich-gelblichen Reflexion des Lichts, siehe Abbildung). Bei Retinoblastompatienten, die später durch andere Symptome klinisch auffällig werden, lässt sich nicht selten auf früheren Photographien bereits das Phänomen der Leukokorie beobachten. Liegt gleichzeitig auf dem Auge auch noch eine tumorbedingte „Blindheit“ (Amaurose) vor, so spricht man auch vom „amaurotischen Katzenauge“.
Neben der Leukokorie ist eine Schielstellung (Strabismus) der Augen das häufigste Symptom. Seltener findet man eine schmerzhafte Rötung des Auges, ein Glaukom (grüner Star), teilweisen Verlust des Sehvermögens, Entzündungen in der Augenhöhle (orbitale Cellulitis), sehr selten eine einseitige Weitstellung der Pupille (unilaterale Mydriasis), Verfärbungen der Iris (Heterochromie), weiße Irisflecken oder Blut in der vorderen Augenkammer (Hyphäma).
Diagnostik
Gängige Untersuchungsmethoden sind insbesondere die Augenspiegelung (Ophthalmoskopie) und die Sonographie. In bestimmten Fällen wird zudem eine Computertomographie oder Magnetresonanztomographie vorgenommen, bei fortgeschrittenem Stadium der Krankheit auch eine Untersuchung von Liquor und Knochenmark, um eine Metastasierung feststellen zu können.
Reese-Ellsworth-Klassifikation
Dieses für das Retinoblastom am meisten verwendete Klassifikationssystem wurde in den 1960er Jahren am Columbia Presbyterian Medical Center entwickelt. Dabei handelt es sich nicht um ein klassisches Staging-System, bei dem Patienten von Gruppe I aus zu den höheren Gruppen fortschreiten. Es wurde vielmehr entworfen, um die Prognose von Tumoren nach perkutaner Strahlentherapie vorauszusagen: je höher die Klassifikation, desto weiter vorne im Auge sitzt ein Tumor und umso schlechter ist der Behandlungserfolg. Mit dem zunehmenden Trend zu anderen Therapien wird dieses Klassifikationssystem zunehmend in Frage gestellt; andere Systeme konnten sich bisher jedoch noch nicht durchsetzen.
- Ia. - Solider Tumor, <4 Diskus-Ø, am oder hinter dem Äquator
- Ib. - Multiple Tumoren ,<4 Diskus-Ø, am oder hinter dem Äquator
- IIa. - Solider Tumor, 4-10 Diskus-Ø, am oder hinter dem Äquator
- IIb. - Multiple Tumoren, 4-10 Diskus-Ø, am oder hinter dem Äquator
- IIIa.- Alle Läsionen vor dem Äquator
- IIIb. - Solider Tumor >10 Diskus-Ø hinter dem Äquator
- IVa. - Multiple Tumoren, >10 Diskus-Ø
- IVb. - Alle Läsionen rostral der Ora serrata
- Va. - Tumor nimmt über die Hälfte der Retina ein
- Vb. - Glaskörperaussaat
Therapie
 Glasauge, Vorder- und Rückseite eines Glasauges
Glasauge, Vorder- und Rückseite eines GlasaugesDie Chance auf Heilung sowie auf Erhalt der Sehfähigkeit ist sehr stark von der Ausbreitung des Tumors im und außerhalb des Auges abhängig. Hauptziel ist in jedem Falle die Rettung des Patienten, was auf eine vollständige Entfernung des Tumors hinausläuft. Wenn es möglich ist, versucht man dabei den Augapfel und möglichst viel von der Sehfähigkeit zu erhalten.
Enukleation
Bei fortgeschrittenem Krankheitsstadium kann der Tumor nur noch durch die Entfernung des betroffenen Auges vollständig beseitigt werden. Bei dieser Enukleation wird das Auge mit einem möglichst großen Teil des Sehnerven (über den eine Ausbreitung von Metastasen ins Gehirn möglich ist) operativ entfernt. Die Augenmuskeln und der übrige Inhalt der Orbita bleiben dabei erhalten. Tumor und Sehnerv werden im Anschluss an die Enukleation histologisch untersucht, um eine eventuelle Metastasierung feststellen zu können.
Im Anschluss an die Entfernung des Augapfels wird ein Implantat in die Augenhöhle eingesetzt, auf das eine Augenprothese aufgesetzt werden kann.
Perkutane Strahlentherapie
Das Retinoblastom reagiert äußerst sensibel auf Bestrahlung. Daher war die perkutane Strahlentherapie (Bestrahlung des Tumors durch die Haut unter Aussparung der Linse) über lange Zeit die Standardtherapie. Allerdings weiß man inzwischen, dass sich das Risiko für Sekundärtumoren durch die Bestrahlung (vor allem beim erblichen Retinoblastom und einer Bestrahlung im ersten Lebensjahr) um den Faktor 3-6 erhöht. Heute verwendet man die perkutane Strahlentherapie vor allem bei Glaskörperaussaat, Metastasierung oder wenn sich eine Chemotherapie als unwirksam oder unverträglich erwiesen hat. Mögliche Nebenwirkungen sind strahlungsbedingte Schädigungen der Bestandteile des Auges wie der Netzhaut (Strahlenretinopathie), des Sehnerven (Strahlenopticusneuropathie und Opticusatrophie), der Linse (Strahlenkatarakt) oder Tränendrüse sowie Störungen des Knochenwachstums.
Brachytherapie
Bei der Brachytherapie werden Strahlenträger (Applikatoren) auf die Lederhaut (Sclera) des Auges aufgenäht und nach Erreichen der erforderlichen Strahlendosis wieder entfernt. Die Applikatoren enthalten kleine Stückchen eines radioaktiven Strahlers wie etwa Ruthenium-106 oder Iod-125, dessen Strahlung recht gezielt auf den Tumor ausgerichtet werden kann. Somit eignet sie sich vor allem für mittelgroße Tumoren abseits der Makula und lokalisierte Glaskörperaussaaten. Mögliche Nebenwirkungen sind Doppelsehen, Trübungen der Linse, Schädigungen der Netzhaut oder des Sehnerven oder Blutungen.
Koagulationstechniken
Hierbei handelt es sich um lokale Therapieverfahren, bei denen der Tumor durch Erhitzung mit einem Laser (Laserkoagulation) oder Vereisung (Kryokoagulation) abgetötet wird. Sie werden bei Patienten mit relativ frühen Stadien und kleinen Tumoren angewendet, bei Rezidiven nach Bestrahlung oder nach Chemoreduktion. Die Tumoren müssen hierfür von Sehnerv, Makula, Aderhaut und größeren Gefäßen entfernt sein, damit diese nicht durch die Behandlung geschädigt werden. Als Komplikationen können Myopie, Schrumpfung der Lederhaut und Schäden an der Netzhaut auftreten.
Chemotherapie
 Retinoscan eines Retinoblastoms vor und während einer Chemotherapie
Retinoscan eines Retinoblastoms vor und während einer ChemotherapieEine adjuvante (unterstützende) Chemotherapie wird in erster Linie nach einer Enukleation durchgeführt, wenn eine Metastasierung vorliegt und Tumorzellen in Vorderkammer, Iris oder Aderhaut oder im entfernten Sehnerv gefunden wurden.
Bei der Chemoreduktion werden große Tumoren im vorderen Teil des Auges durch eine Chemotherapie verkleinert, bevor sie mit lokalen Therapien (Brachytherapie, Laser- oder Kryokoagulation) weiter behandelt werden.
Eine Hochdosis-Chemotherapie wird nur bei starker Metastasierung mit schlechter Prognose durchgeführt.
Neben den üblichen Nebenwirkungen einer Chemotherapie ist das Auftreten von Chemotherapie-Resistenz problematisch, durch die der Tumor nach einer Weile das Wachstum wieder aufnimmt.
Thermochemotherapie
Bei der Thermochemotherapie macht man sich den Effekt zunutze, dass eine lokale Erwärmung des Tumors den Effekt der Behandlung mit einem Chemotherapeutikum (in der Regel Carboplatin) wesentlich verbessert. Sie wird vor allem bei kleineren Tumoren im hinteren Teil des Auges angewendet. Mögliche Komplikationen sind Trübungen des Glaskörpers und Schäden an Netzhaut und Iris.
Neue Therapieverfahren
Alle aktuellen Therapieverfahren sind mit einer Reihe von Nachteilen verbunden: die Enukleation führt zum Verlust eines Auges, die perkutane Strahlentherapie erhöht das Risiko von Sekundärtumoren, die Chemotherapie ist mit erheblichen Nebenwirkungen verbunden und ist durch das Auftreten von Resistenzen nur begrenzt wirksam, Laserkoagulation und Kryokoagulation führen zum Verlust von Netzhautarealen und damit zu Einschränkungen des Sehvermögens. Trotz einer geringen Mortalität ist das Retinoblastom somit mit einer hohen Morbidität verbunden. Neuartige Therapieverfahren wie die Thermochemotherapie oder Photodynamische Therapie sollen die Morbidität weiter senken.
Geschichtliches
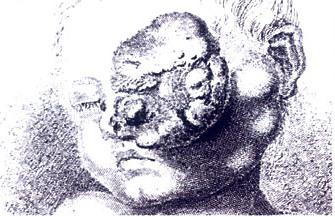
 Wardrops Illustration eines fortgeschrittenen Retinoblastoms, das bereits aus der Augenhöhle herauswächst
Wardrops Illustration eines fortgeschrittenen Retinoblastoms, das bereits aus der Augenhöhle herauswächstDie erste Beschreibung eines Retinoblastoms wird dem Leidener Anatomen Pieter Pauw, genannt Petrus Parwius zugeschrieben, der 1597 in seinen "Observationes Anatomicae Selectiores" im Kapitel "Tumor oculorum" die Obduktion eines dreijährigen Knaben beschreibt, der an einem großen Tumor des linken Auges verstorben war. William Hey beschreibt 1805 in seinen "Practical Observations in Surgery" eingehend einen "Fungus haematodes", also Blutschwamm. Die detaillierteste Untersuchung findet man in den “Observations on the Fungus Haematodes or Soft Cancer” von James Wardrop (1809), der 17 Fälle beschreibt und erstmals die Entstehung in der Netzhaut und die Ausbreitung über den Sehnerv (N. opticus) beschreibt. Als erster empfiehlt er die Entfernung des Auges. 1836 können Langenbech, Robin und Nystin erstmals die Entstehung in der Netzhaut mikroskopisch beweisen. Rudolf Virchow bezeichnet den Tumor 1864 als Glioma retinae. 1868 erkennt Albrecht von Graefe, dass es sich um eine erbliche Krankheit handelt. Simon Flexner (1891) und Hugo Wintersteiner (1897) beschreiben das typische rosettenförmige Wachstum (Flexner-Wintersteiner-Rosetten) und erkennen die Beziehung zu Stäbchen und Zapfen der Netzhaut. Der Begriff "Retinoblastom" wird von Fredrick Herman Verhoeff 1926 geprägt, der die Herkunft aus noch undifferenzierten Netzhautzellen, den Retinoblasten, erkennt. 1971 dient das Retinoblastom als Modellsystem für die "Two-Hit"-Hypothese von Alfred G. Knudson, und 1987 wird das "Retinoblastoma susceptibility gene" von Lee sequenziert.
Literatur
- Flexner S. A peculiar glioma (neuroepithelioma?) of the retina. Johns Hopkins Hosp Bull 1891;2:115–19
- Lee WH, Bookstein R, Hong F, Young LJ, Shew JY, Lee EY. Human retinoblastoma susceptibility gene: cloning, identification, and sequence. Science. 1987 Mar 13;235(4794):1394-9 (PDF) [1]. PMID 3823889
- Chintagumpala M, Chevez-Barrios P, Paysse EA, Plon SE, Hurwitz R. Retinoblastoma: review of current management. Oncologist. 2007 Oct;12(10):1237-46. Review. PMID 17962617
- Melamud A, Palekar R, Singh A. Retinoblastoma. Am Fam Physician. 2006 Mar 15;73(6):1039-44. Review. PMID 16570739
- Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 1971 Apr;68(4):820–3. PMID 5279523
- Bornfeld et al: Perspektiven der Ophthalmoonkologie. Dtsch Arztebl 2004; 101(38): A-2526 / B-2130 / C-2049
Einzelnachweise
Weblinks
- Diagnose und Therapie des Retinoblastoms (PDF) (609 kB)
- Genetik des Retinoblastoms (PDF) (596 kB)
- Kinder-Augen-Krebs-Stiftung. Abgerufen am 19. Juni 2010 (Aktuelle Informationen über Krankheitsbild, Forschung und Vorsorge).
- Retinoblastom-Seite der Deutschen Krebsgesellschaft e. V.
- Informationen auf Kinderkrebsinfo.de (Gesellschaft für Pädiatrische Onkologie und Hämatologie)

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Krebserkrankung
- Kinderonkologie
- Krankheitsbild in der Augenheilkunde




Wikimedia Foundation.