- Bence-Jones Plasmozytom
-
Klassifikation nach ICD-10 C90.0 Plasmozytom (Multiples Myelom) ICD-10 online (WHO-Version 2006) Ein multiples Myelom (syn. Plasmozytom, Kahler-Krankheit nach Otto Kahler, Huppert-Krankheit, sog. monoklonale Gammopathie mit pathologischer Produktion von Immunglobulinen) ist eine Krebserkrankung des Knochenmarks. Sie ist gekennzeichnet durch bösartige Vermehrung Antikörper-produzierender Zellen, der Plasmazellen. Die entarteten Plasmazellen produzieren in der Regel Antikörper oder Bruchstücke davon. Da alle malignen Plasmazellen von einer gemeinsamen Vorläuferzelle abstammen, sind sie genetisch identisch (Zellklon) und produzieren identische (= monoklonale) Antikörper.
Das Ausmaß der Bösartigkeit (der Grad der Malignität) kann sehr unterschiedlich sein und reicht von Krebsvorstufen über nur langsam voranschreitende Krankheitsverläufe bis zu hochmalignen, ohne Behandlung schnell zum Tod führenden Erkrankungen.
Krankheitssymptome entstehen entweder durch das bösartige Wachstum der Plasmazellen oder durch die Eigenschaften der gebildeten Antikörper oder Antikörperbruchstücke. Das Wachstum der Plasmazellen führt zu Knochenschmerzen und Auflösung der Knochen bis zu spontanen Knochenbrüchen, Anstieg des aus dem Knochen gelösten Calciums im Blut und Abnahme der im Knochenmark gebildeten roten Blutkörperchen. Die im Übermaß produzierten und oftmals abnormalen Antikörper können durch Ablagerung im Gewebe zu Funktionsstörungen vieler Organe, zu Nierenversagen und zur Beeinträchtigung der Durchblutung führen.
Die Diagnose wird durch Blutuntersuchung, Röntgen der großen Knochen und Knochenmarkpunktion gestellt.
Im Frühstadium der Erkrankung wird der Krankheitsverlauf nur beobachtet. Treten Symptome und Komplikationen auf, können Betroffene mit Chemotherapie, Medikamenten, die das Immunsystem beeinflussen, Medikamenten, die die Knochenauflösung hemmen, und Knochenmarktransplantation behandelt werden.
Nach Auftreten von Komplikationen beträgt die Lebenserwartung ohne Behandlung im Mittel 6 Monate, mit Chemotherapie 3 Jahre und nach Knochenmarkstransplantation 5 Jahre.
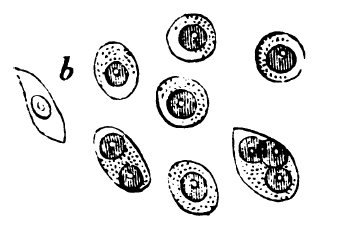
_MG_stain.jpg) Knochenmarkausstrich bei Multiplem Myelom. Färbung nach May-Grünwald-Giemsa. Vermehrung von Plasmazellen (Große ovale Zellen mit breitem Zytoplasma und exzentrisch gelegenem Zellkern).
Knochenmarkausstrich bei Multiplem Myelom. Färbung nach May-Grünwald-Giemsa. Vermehrung von Plasmazellen (Große ovale Zellen mit breitem Zytoplasma und exzentrisch gelegenem Zellkern).Inhaltsverzeichnis
Terminologie
Das Plasmozytom zählt zu den aggressiven B-Zell-Non-Hodgkin-Lymphomen. Das Spektrum der monoklonalen Plasmazellerkrankungen reicht mit steigender Bösartigkeit (Malignität) von der Krebsvorstufe (Präkanzerose) 'Monoklonale Gammopathie unklarer Signifikanz' (MGUS) über das langsam progrediente 'indolente Myelom (smoldering Myeloma)' bis zum symptomatischen multiplen Myelom. Der Begriff 'Plasmozytom' steht dabei für einen solitären intramedullären (im Knochenmark gelegenen) oder ein oder mehrere extramedullär gelegene Herde maligner Plasmazellen, von 'multiplem Myelom' spricht man, sobald mehr als ein intramedullärer Herd vorhanden ist. Der besseren Übersichtlichkeit halber verwendet dieser Artikel die Begriffe "Multiples Myelom" und "Plasmozytom" synonym.
Epidemiologie
Die Inzidenz des Plasmozytoms liegt bei etwa 4 Neuerkrankungen/100.000 pro Jahr. Es ist eine Erkrankung des höheren Lebensalters, das mediane Alter bei Diagnosestellung liegt bei 66 Jahren, nur 2% der Patienten sind jünger als 40 Jahre. Es tritt bei Männern etwas häufiger als bei Frauen auf, bei Afroamerikanern kommt die Erkrankung im Vergleich zur weißen US-amerikanischen Bevölkerung etwa doppelt so häufig vor.
Risikofaktoren für die Entstehung eines Multiplen Myeloms
Die pathogenetische Ursache des Plasmozytoms ist weitgehend unbekannt. Diskutiert wird der Einfluss radioaktiver Strahlung, zum Teil wurden verschiedene genetische Translokationen beschrieben, deren Einfluss aber noch nicht geklärt sind. Mutationen in Onkogenen wurden ebenfalls beobachtet. Interessanterweise scheinen Häufungen bei bestimmten Berufsgruppen (Wald- und Lederarbeiter, Landwirte, Friseure) zu bestehen.
Pathogenese
Der Übergang von einer monoklonalen Gammopathie unklarer Signifikanz zum aktiven Myelom verläuft in vielen aufeinanderfolgenden Schritten und kann wenige Monate bis mehrere Jahrzehnte dauern[1].
Die maligne Entartung der Plasmozytom-Zellen findet meist außerhalb des Knochenmarks in Keimzentren peripherer lymphatischer Organe statt. Die B-Zellen, die in diese Keimzentren eintreten, haben schon die ersten Differenzierungschritte (V-D-J-Rearrangements und Isotyp-switch) hinter sich. In diesem Stadium treten die genetischen Veränderungen ein, die letztlich zur Entstehung des Plasmozytoms führen.
Bei den meisten Patienten beobachtet man Translokationen, die bewirken, dass ein Onkogen unter die Kontrolle eines regulatorischen Gens gelangt und somit stark aktiviert wird. Beim Plasmozytom ist dies am häufigsten (ca. 80%) das Immunglobulin-Enhancer-Gen auf Chromosom 14q31. Häufige Partner dieser Translokation sind Teile des Chromosoms 4 (4p16.3; Fibroblast Growth Factor Receptor), 6 (6p21; Cyclin D3), 11 (Bcl-1, Cycin D1), 20 (20p11; maf8) und 16 (16q23; C-maf). Selten findet man 8q24 (c-myc) und noch seltener 18q21 (bcl-2), 11q23 (MLL-1) sowie 20q11 (maf B). Im weiteren Verlauf der Erkrankung treten noch andere genetische Veränderungen hinzu. Die unterschiedlichen genetischen Veränderungen gehen mit unterschiedlichen Krankheitsverläufen einher und definieren vielleicht eigene Krankheiten.
Der Nachweis bestimmter genetischer Veränderungen in Plasmozytomzellen eines Patienten lässt in manchen Fällen Rückschlüsse auf die Prognose zu. So weiß man beispielsweise, dass eine Deletion von Chromosom 13 (Monosomie 13) ein Hinweis auf eine kürzere Überlebenszeit sein kann.
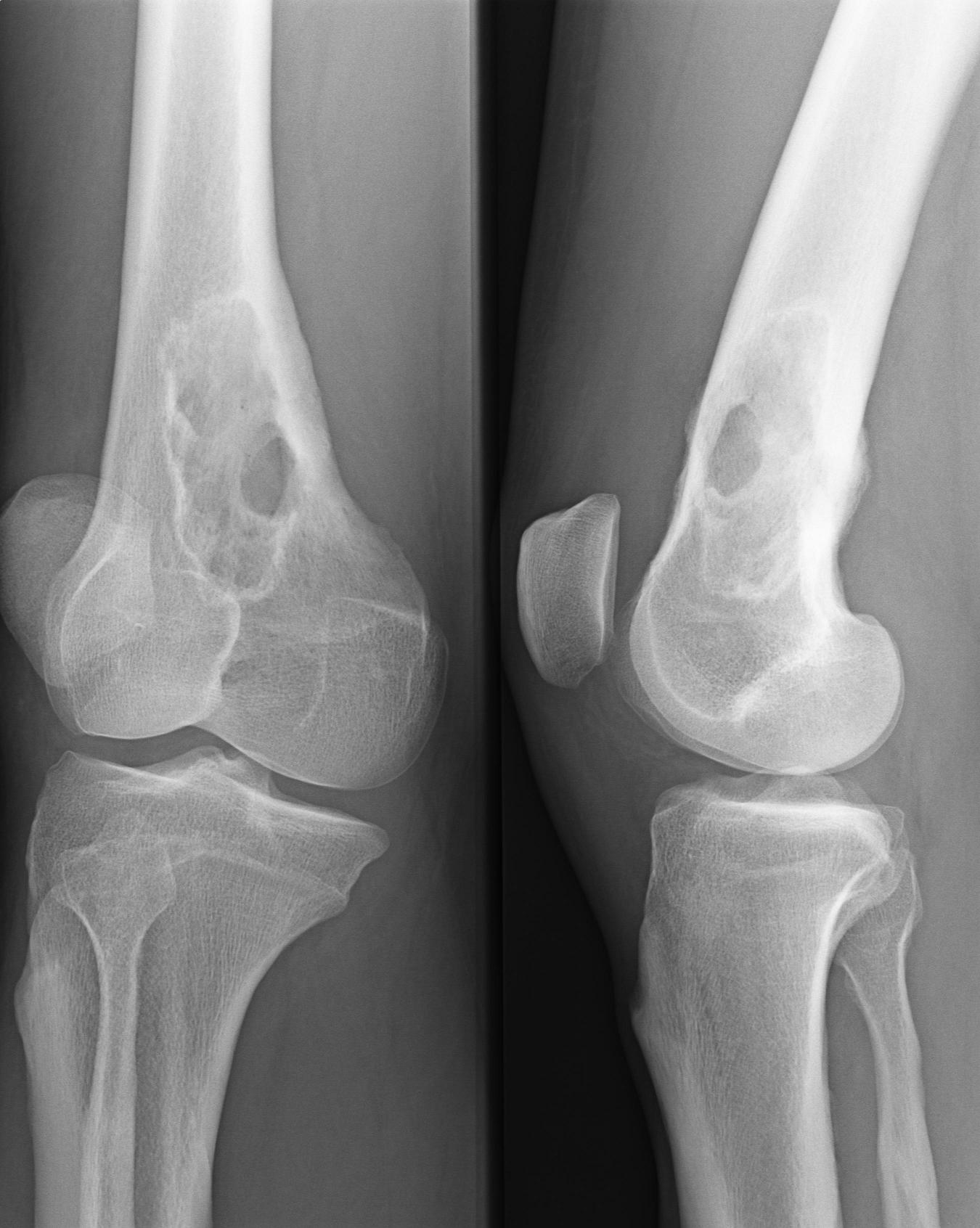
Nach der klonalen Vermehrung einer entarteten Plasmazelle kommt es zur Infiltration des Knochenmarks mit folgender Zerstörung des Knochens und Verdrängung der normalen Hämatopoese (Blutbildung). Hierbei scheiden die malignen Zellen Wachstumsfaktoren und Zytokine aus, welche die Osteoklasten aktivieren (u.a. OAF), was letztlich zu einem Knochenabbau führt. Dies führt zu einer allgemeinen Osteoporose sowie zu den charakteristischen, im Röntgenbild sichtbaren Knochenschäden, die im Gegensatz zu Knochenmetastasen wie ausgestanzt dargestellt werden. Es fehlt der bei Knochenmetastasen sonst vorhandene Saum aus reaktiven Zellen („osteoblastischer Randsaum“). Dies ist auch der Grund dafür, dass sich die Knochenveränderungen bei einem Plasmozytom nicht im Knochenszintigramm darstellen können. Insgesamt entwickeln nur etwa 60% der Patienten Knochenveränderungen.
Die malignen Zellen bilden Antikörper oder Antikörperteile (sogenannte Leichtketten), welche sich im Körper anreichern und für viele Symptome und Komplikationen der Erkrankung verantwortlich sind.
Entsprechend dem gebildeten Antikörper werden folgende Typen unterschieden:
- IgG - Plasmozytom (>50 Prozent)
- IgA - Plasmozytom (25 Prozent)
- IgD - Plasmozytom (selten)
- Leichtkettenplasmozytom (Bence-Jones-Plasmozytom/Leichtkettenkrankheit)
- κ - Ketten - Plasmozytom
- λ - Ketten - Plasmozytom
Symptome
Bei einigen Patienten wird die Erkrankung zufällig im Rahmen einer Blutuntersuchung anhand einer auffälligen Serumelektrophorese diagnostiziert.
Bei den meisten Patienten treten jedoch Symptome und Komplikationen auf:
- Allgemeinsymptome wie Schwäche und Gewichtsverlust
- Verminderung der Knochenmarksfunktion (Knochenmarkinsuffizienz) mit Blutarmut (Anämie), Verminderung der weißen Blutkörperchen (Leukopenie) und/oder der Blutplättchen (Thrombozytopenie)
- oft fällt ein sehr starker Abbau der Knochensubstanz (Osteoporose) als erstes Symptom auf, "Osteoporose" wird als häufige Fehldiagnose gestellt
- Infektanfälligkeit durch Antikörpermangel (die vom Plasmozytom gebildeten Antikörper sind ohne Funktion) und Mangel an weissen Blutkörperchen (Leukopenie).
- Knochenschmerzen und Knochenbrüche durch Auflösung des Knochengewebes (Osteolysen). Osteolysen im Schädelknochen stellen sich im Röntgenbild als "Schrotschussschädel" dar.
- Hypercalciämie (erhöhter Calciumspiegel im Blut) kann zu Schwäche und Nierenschäden führen
- Die überschießende Bildung von Immunglobulinen kann die Viskosität des Serums erhöhen. Dies kann zu Störungen der Mikrozirkulation führen, die sich in Kopfschmerzen, Benommenheit, Schwindel, Nystagmus, Hör- und Sehstörungen, Schläfrigkeit, Koma und Krampfanfällen äußern (Hyperviskositätssyndrom).
- Die Ausscheidung von Leichtketten im Urin kann die Nierenfunktion auf unterschiedliche Weise beeinträchtigen[2]:
- In den Nierenkanälchen kann es in Gegenwart von Tamm-Horsfall-Protein zu Ausfällungen der Leichtketten in Form von Eiweißzylindern (Cast-Nephropathie) kommen. Diese Ausfällungen wirken direkt toxisch auf die Zellen der Nierenkanälchen und können zu einem raschen Verlust der Nierenfunktion (akutes Nierenversagen) führen (klassische Myelomniere).
- Ablagerung von Leichtketten in den Basalmembranen von Nierenkörperchen und Nierenkanälchen kann zur Leichtketten-Ablagerungs-Krankheit (light-chain deposition disease) führen. Diese äußert sich in einer stark vermehrten Ausscheidung von Eiweiß im Urin (nephrotisches Syndrom) und führt zu einem chronischen Nierenfunktionsverlust.
- Die Leichtketten können zur Funktionsstörungen der Zellen im Hauptstück der Nierenkanälchen führen (Fanconi-Syndrom). Diese Funktionsstörungen äußern sich in einer verminderten Ausscheidung von Säuren (Azidose) sowie einem vermehrten Verlust von Phosphat, Glukose, Harnsäure und Aminosäuren.
- Durch Zusammenlagerung von Leichtketten im Bereich der β-Faltblatt-Strukturen können sich Fibrillen bilden, feine langgestreckte Strukturen, die sich in Form von Amyloid in Nierengewebe und Nierengefäßen ablagern. Folge sind wie bei der Leichtketten-Ablagerungs-Krankheit erhöhte Eiweißausscheidung und chronischer Nierenfunktionsverlust.
Diagnose
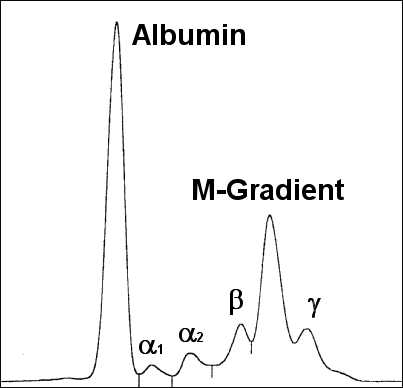
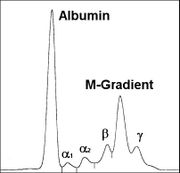 Monoklonale Gammopathie bei IgA-Plasmozytom
Monoklonale Gammopathie bei IgA-Plasmozytom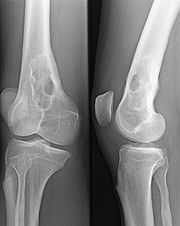 Osteolyse durch ein Plasmozytom - Oberschenkelknochen
Osteolyse durch ein Plasmozytom - OberschenkelknochenEinen Hinweis auf das Vorliegen eines Plasmozytoms gibt der Nachweis einer monoklonalen Gammopathie im Blut und/oder einer Leichtkettenausscheidung im Urin durch Serumelektrophorese und Immunelektrophorese oder Immunfixations-Elektrophorese. Man findet monoklonale Immunglobuline aber auch bei MGUS (monoklonae Gammopathie unklarer Signifikanz) sowie ebenfalls bei reaktiven Knochenmarkveränderungen. Einen wichtigen Hinweis auf das Vorliegen eines Plasmozytoms geben aber deutlich erhöhte Serumkonzentrationen (IgG > 3,5 g/dl, IgA > 2,0 g/dl) oder der Nachweis einer erhöhten Konzentration von Leichtketten im Urin (> 1,0 g/24h).
Die Knochenmarkspunktion zeigt eine Infiltration durch Plasmazellen. Diese können manchmal eine atypische Form aufweisen, häufig gibt aber nur der gesteigerte Anteil an Plasmazellen einen Hinweis auf die Erkrankung. Man findet auch bei Entzündungen oder reaktiven Veränderungen im Knochenmark Plasmazellanteile bis zu 30%.
Findet man zusätzlich zu einem Paraprotein und zur Plasmazell-Vermehrung im Knochenmark noch typische Komplikationen wie eingeschränkte Nierenfunktion, Hyperkalziämie, Anämie oder typische Knochenveränderungen, kann die Diagnose des Plasmozytoms als gesichert gelten.
Osteolysen lassen sich mit konventioneller Röntgen-Untersuchung der Knochen nachweisen. Das Skelettszintigramm zeigt oft keine Aktivitätsänderung im osteolytischen Bereich, ist hier also nicht verlässlich. Ein Knochenszintigramm eignet sich aber zum Nachweis von Knochenbrüchen ("pathologische Fraktur") aufgrund der Infiltration durch das Plasmozytom. Zur Frühdiagnose einer Plasmozytominfiltration in den Knochen kann das MRT beitragen, hier zeigt sich auch die Reaktion im Weichteilmantel, bevor es zu Osteolysen gekommen ist.
Typische weitere Laborveränderungen sind eine sehr stark erhöhte BSG (Blutkörperchensenkungsgeschwindigkeit) sowie eine erhöhte LDH, ein erhöhtes CRP und ein erhöhtes β2-Mikroglobulin.
Differenzialdiagnostisch abzugrenzen ist neben reaktiven Knochenmarkveränderungen das "smouldering Myeloma" und anderen malignen Lymphomen die eher harmlose MGUS (monoklonale Gammopathie unklarer Signifikanz).
Diagnostische Kriterien
Hauptkriterien
- monoklonale Paraproteinämie:
- Serum: IgG > 35 g/l, IgA > 20 g/l
- Urin: Bence-Jones-Proteinurie > 1 g/24h
- Knochenmarkinfiltration von > 30% Plasmazellen
- histologischer Plasmozytomnachweis in sonstigen Biopsien
Nebenkriterien
- Knochenmarkinfiltration von >10% Plasmazellen
- monoklonale Paraproteinämie in geringerer Konzentration als im Hauptkriterium
- Osteolysen
- Antikörpermangel: IgM < 0.5 g/l, IgA < 1 g/l, IgG < 6 g/l
Stadieneinteilung
Die Einteilung nach Durie und Salmon unterscheidet 3 Stadien:
Stadium Merkmale Stadium I Hämoglobin > 10 g/dl
Kalzium im Serum normal
maximal eine Osteolyse
IgG < 5 g/dl bzw. IgA < 3 g/dl
Leichtkettenausscheidung im Urin < 4 g/24hStadium II zwischen Stadium I und III Stadium III mindestens eines der folgenden Kriterien:
Hämoglobin < 8,5 g/dl
erhöhtes Kalzium
mehrere Osteolysen
IgG > 7 g/dl
IgA > 5 g/dl
Leichtkettenausscheidung im Urin > 12 g/24hZusatz
A
B
falls Kreatinin < 2 mg/dl (177µmol/l)
falls Kreatinin ≥ 2 mg/dl (177µmol/l)Therapie
Im Stadium I wird nur bei Gefahr der Nierenschädigung durch die falschen Antikörper oder deren Leichtketten therapiert. Im Stadium II wird eine zyklische Chemotherapie durchgeführt. Das klassische Therapieschema nach Alexanian ist eine Kombination von Melphalan und Prednison. Alternativ zu Melphalan kann auch Cyclophosphamid oder Bendamustin eingesetzt werden. Bei jüngeren Patienten, bei denen eine autologe Stammzelltransplantation geplant ist, wird auf Melphalan zunächst verzichtet und mit VID (Vincristin, Idarubicin, Dexamethason) oder VAD (Vincristin, Adriamycin, Dexamethason) behandelt. Eine neue Therapieoption ist die Behandlung mit dem Proteasominhibitor Bortezomib. Bei einem 50prozentigem Rückgang der Immunglobulinkonzentration kann von Remission gesprochen werden. Das Fortschreiten der Osteolysen kann durch die Gabe von Bisphosphonaten verzögert werden. Im Stadium III kann bei Knochenschmerzen und drohenden Frakturen eine lokalisierte Bestrahlung durchgeführt werden. Die Frakturpunkte werden operativ fixiert. Bei Infektanfälligkeit werden Antikörper gegeben, bei Anämie Bluttransfusionen durchgeführt. Das Hyperviskositätssyndrom kann kurzzeitig durch eine Plasmapherese behoben werden.
Seit 2001 steht Thalidomid als sogenanntes Arzneimittel für seltene Leiden (Orphan-Arzneimittel) für die Behandlung des multiplen Myeloms zur Verfügung. In Europa vertreibt Pharmion Thalidomid nach dem sogenannten „Compassionate Use“-Prinzip, wobei einige Krankenkassen die Therapiekosten zeitweilig nicht ohne weiteres übernehmen wollten. In Kombination mit der Melphalan/Prednison-Therapie verbessert sich die Überlebensrate bei älteren Patienten wesentlich.[3]
Für die Behandlung von Patienten, die mindestens eine vorausgegangene Therapie erhalten haben, wurde von der Europäischen Arzneimittelagentur (EMEA) im Juli 2007 die perorale Lenalidomid Darreichungsform (Revlimid® Hartkapseln, Celgene) in Kombination mit Dexamethason zugelassen. Lenalidomid gehört zur Gruppe der immunmodulatory drugs (IMiDs). In der Struktur ähnelt Lenalidomid dem Thalidomid, die biologische Aktivität ist aber stärker. In Verbindung mit Dexamethason ist es bei Rückfällen oder Therapieversagen effektiver als eine Monotherapie mit Dexamethason.[4]
Ohne Behandlung liegt das Überleben eines Betroffenen eines aktiven Myeloms im Median bei 6 Monaten. Die Behandlung mit Melphalan/Prednison verlängert das mediane Überleben auf 3 Jahre. Hochdosis-Chemotherapie mit autologer Stammzelltransplantation verbessert das mediane Überleben weiter auf 5 Jahre.
Geschichte des Multiplen Myeloms
Das Multiple Myelom ist keine Erscheinung der Neuzeit, bereits seit vielen Jahrhunderten werden Menschen von dieser Erkrankung heimgesucht. So weisen schon Knochenfunde aus dem 2. Jahrhundert v. Chr. die typischen zerstörerischen Merkmale der Erkrankung auf [5].
Historischer Fallbericht: Thomas McBean (1850)
Der erste gut dokumentierte Fall eines Multiplen Myeloms geht auf Macintyre zurück[6][7]. Er beschrieb die Krankheitsgeschichte des englischen Gemischtwarenhändlers Thomas A. McBean, welcher sich 1845 im Alter von 45 Jahren in seiner Arztpraxis vorgestellt hatte. McBean beklagte, dass etwas mit seinem Urin nicht stimmte – er verspürte häufig Harndrang, und sein „Leibkleid wurde durch den Urin ganz steif“, obwohl er keinen Ausfluss aus der Harnröhre bemerkt hatte. Außerdem litt der Patient an ungewöhnlicher Schwäche und Ausgezehrtheit, während eines Spaziergangs habe er dann das Gefühl gehabt, „etwas im Brustkorb knackte oder gab nach“, und McBean stürzte und konnte sich „wegen starker Schmerzen für einige Minuten nicht mehr erheben“. Doktor Macintyre behandelte den Patienten mit einer Bandage des Brustkorbes („strenghening plasters“) und verordnete körperliche Schonung[6]. Einen Monat später hatte der Patient erneut starke Schmerzen, woraufhin man wiederholte Aderlässe, Blutegelbehandlungen und Schröpfkegel einsetzte, was jedoch keine dauerhafte Linderung brachte, so dass sich McBean bei einem anderen Arzt, Dr. Watson, vorstellte. Dieser begann eine Behandlung mit Eisen und Chinin, was zu einer erstaunlichen Besserung führte, die ein halbes Jahr anhielt. Im Oktober 1845 erlitt der Patient jedoch starke Schmerzen an der Wirbelsäule und des Ischias, welche sich auch durch den Einsatz von warmen Bädern, Kampferpuder und –salbe nicht besserten. Dr. Macintyre diagnostizierte außerdem Ödeme am Körper von McBean und untersuchte daher den Urin des Patienten. Dieser war dunkel und flockte bei Erhitzung aus („abound in animal matter“). Fast gleichzeitig schickte Dr. Watson mit der Frage „What is it?“ eine Urinprobe an den Arzt und Chemiker Bence Jones, welcher Proteine im Urin von McBean - und anderer Patienten mit ähnlichen Beschwerden - feststellte und diese charakterisierte[8]. Der Zustand von McBean verschlechterte sich im Verlaufe des Jahres 1845 rapide, er hatte starke Schmerzen und konnte das Bett nicht mehr verlassen. Am 1. Januar 1846 verstarb McBean schließlich, in seiner Todesurkunde wurde „Atrophie durch Albuminurie“ als Todesursache festgehalten[7].
 Historischer Holzschnitt der histologischen Befunde beim Multiplen Myelom[9]
Historischer Holzschnitt der histologischen Befunde beim Multiplen Myelom[9]Die Obduktion, bei der die Doktoren Macintyre, Watson und Jones anwesend waren, zeigte Knochen, die „leicht mit dem Messer zu schneiden waren und einfach brachen“. Die Rippen zerbröckelten förmlich und enthielten eine blutrote, gelatineartige und ölige Masse. Auch die gesamte Wirbelsäule war von ähnlicher Beschaffenheit. Becken-, Oberarm- und Oberschenkelknochen „widerstanden jedoch jeden Versuch, sie mit der Hand zu brechen“. Herz, Lunge und Leber wurden als weitgehend unauffällig beschrieben[6]. John Dalrymple, Chirurg und Mitglied der mikroskopischen Fachgesellschaft, untersuchte zwei Lendenwirbel und eine Rippe von McBean. Er stellte Löcher im Knochen des Patienten fest, welche mit einer roten, gelartigen Masse gefüllt waren. Diese untersuchte er unter dem Mikroskop und fand große, gleichförmig aussehende runde bis ovale Zellen, teilweise mit mehreren Nuclei. Die nach Zeichnungen von Dalrymple angefertigten Holzschnitte zeigen die noch heute gültigen Kriterien für Myelomzellen[7].
Literatur
- Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med 2004;351:1860-73. (PMID 15509819)
- Richardson PG, Mitsiades C, Hideshima T, Anderson KC. Proteasome Inhibition in the Treatment of Cancer. Cell Cycle 2005;4(2) (PMID 15655370)
- Barlogie B, Shaughnessy J, Tricot G et al. Treatment of multiple myeloma. Blood 2004; 103:20-32. (PMID 12969978)
- Ludwig H, Schmoll HJ: Multiples Myelom. in: Schmoll, Höffken, Possinger: Kompendium Internistische Onkologie. Therapiekonzepte maligner Tumoren. 4. Auflage, Springer Verlag Heidelberg, 2006. Seite 3165–3206. ISBN 3-540-20657-4
Einzelnachweise
- ↑ Singhal, Seema, Mehta, Jayesh: Multiple Myeloma. Clin J Am Soc Nephrol 2006 1: 1322-1330
- ↑ Dember, Laura M.: Light Chains, Casts, Sheets and Fibrils: Monoclonal Immunoglobulin Diseases and Immunotactoid/Fibrillary Glomerulopathy. Clin J Am Soc Nephrol 2006 1: 1320-1321
- ↑ Thierry Facon and others.: „Melphalan and prednisone plus thalidomide versus melphalan and prednisone alone or reduced-intensity autologous stem cell transplantation in elderly patients with multiple myeloma (IFM 99–06): a randomised trial..“ Lancet 2007; 370: S. 1209 Abstract
- ↑ Dimopoulos, M. et al.: „Lenalidomide plus Dexamethasone for Relapsed or Refractory Multiple Myeloma.“ N Engl J Med 2007; 357: S. 2123-2132 Abstract
- ↑ Morse, D, Dailey, RC, Bunn, J: „Prehistoric multiple myeloma.“ Bulletin of the New York Academy of Medicine 1974; 50: S. 447-458 Abstract
- ↑ a b c Macintyre, W: „Case of mollities and fragilitas ossium, accompanied with urine strongly charged with animal matter.“ Medical and Chirurgical Transactions of London 1850; 33: S. 211-232
- ↑ a b c Kyle RA: „Multiple myeloma: an odyssey of discovery.“ British Journal of Haematology 2000; 111(4): S. 1035-1044 Abstract
- ↑ Bence Jones, H: „On a new substance occurring in the urine of a patient with mollities ossium.“ Philosophical Transactions of the Royal Society of London (Biology) 1848: S. 55-62
- ↑ Dalrymple, J: „On the microscopical character of mollities ossium.“ Dublin Quarterly Journal of Medical Science 1846; 2: S. 85-95
Weblinks
- Arbeitsgemeinschaft Plasmozytom / Multiples Myelom (APMM)
- Informationen der Deutschen Krebshilfe
- Informations- und Kommunikationsplattform für Ärzte, Betroffene und Angehörige
- Myelom Kontaktgruppe Schweiz
- Multiples Myelom Hilfe Österreich
- Multiples Myelom Pathologie - Bilddatenbank Pathopic der Universität Basel (PathoPic - Anleitung)
- International Myeloma Foundation englischsprachige Seite der "International Myeloma Foundation". Link zu einem deutschsprachigen verständlichen Patientenhandbuch. Aktuellere Informationen sind meist im englischsprachigen Patientenhandbuch eingearbeitet.

Bitte beachte den Hinweis zu Gesundheitsthemen!
_MG_stain.jpg)
{kind=link}
Wikimedia Foundation.