- Drepanozytose
-
Klassifikation nach ICD-10 D57.0 Sichelzellenanämie mit Krisen Hb-SS-Krankheit mit Krisen
D57.1 Sichelzellenanämie ohne Krisen D57.2 Doppelt heterozygote Sichelzellenkrankheit D57.3 Sichelzellen-Erbanlage Heterozygotes Hämoglobin S
D57.8 Sonstige Sichelzellenkrankheiten ICD-10 online (WHO-Version 2006) Die Sichelzellenanämie (med.: Drepanozytose), auch Sichelzellanämie (englisch: sickle cell anemia) ist eine erbliche Erkrankung der roten Blutkörperchen (Erythrozyten). Die Betroffenen bilden einen abnormen roten Blutfarbstoff (Hämoglobin), wodurch sich ihre roten Blutzellen insbesondere bei Sauerstoffarmut zu sichelförmigen Gebilden verformen. Die solchermaßen veränderten Erythrozyten (rote Blutkörperchen) werden vor allem in der Milz als krank erkannt und abgebaut (Hämolyse). Andererseits sind Betroffene gegen Malaria resistent, wodurch diese Eigenschaft in Malariagebieten sehr verbreitet ist.
Inhaltsverzeichnis
Merkmale
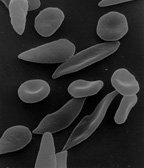 sichelförmige rote Blutkörperchen
sichelförmige rote BlutkörperchenDer Verlust roter Blutkörperchen (Anämie) und rezidivierende Durchblutungsstörungen führen zu starken Schmerzen und multiplen Organveränderungen sowie -schäden: Milzschwellung, Lungenentzündung, Herz- und Nierenversagen. Die Lebenserwartung ist vermindert.
Nur homozygote Träger des Sichelzellgens zeigen diese starke Ausprägung der Krankheit, bei der das gesamte Hämoglobin abnormenes Sichelzellhämoglobin (irreguläres Hämoglobin) ist. In heterozygoten Trägern ist nur etwa 1 % aller Erythrozyten deformiert. Die Symptome verschlimmern sich erheblich, wenn die Patienten körperlich stark aktiv sind oder sich in großen Höhen befinden. Dies liegt daran, dass sich die Sichelform der Erythrozyten bei niedrigem Sauerstoffpartialdruck bildet, weil unter diesen Bedingungen das Hämoglobin faserig ausfällt (die Löslichkeit von Hämoglobin bei Sichelzellanämie ist 25-fach geringer als die Löslichkeit von normalem Hämoglobin).
Etwa ab dem 6. Lebensmonat, wenn der Abbau des fetalen Hämoglobins bereits weit fortgeschritten ist, können erstmals Symptome auftreten. Sie äußern sich dann meist in einer sogenannten Sichelzellkrise: durch äußere Einflüsse, wie z.B. Anstrengung, sinkt der Sauerstoffpartialdruck im Blut, die Sichelzellen werden hämolytisch.
Eine Glomerulopathie mit Hyperfiltration tritt bei bis zu einem Drittel der Patienten mit homozygotem Phänotyp schon in der Kindheit auf. Bei bis zu einem Drittel der Patienten kommt es in den ersten Lebensjahrzehnten zum Auftreten einer Proteinurie, in 5 % zu einem terminalen Nierenversagen. [1]
Ursache
Bei der Sichelzellenanämie ist an der Position 6 der β-Untereinheit des Hämoglobins die Aminosäure Glutaminsäure durch Valin ersetzt. Die betroffenen Erythrocyten verformen sich bei abnehmendem Sauerstoffpartialdruck sichelförmig, verfangen sich leicht in den Kapillaren und lysieren überdies sehr schnell. Das Sichelzellenhämoglobin wird als HbS bezeichnet im Gegensatz zum HbA, dem normalen Hämoglobin des erwachsenen Menschen. Heterozygote Träger des Merkmals bilden neben dem HbS auch HbA; dieses reicht aus, um die Funktion der Erythrocyten bei diesen Menschen weitgehend aufrecht zu erhalten.
Genetik
Sichelzellenanämie ist ein autosomal-kodominantes Erbleiden.
- Das Erbgut eines Gesunden enthält die beiden unvollständig dominanten (Auf molekularem Niveau sind A und a codominant) Allele (AA) für das Hämoglobin A. Seine roten Blutkörperchen sind stets elastisch.
- Ein Überträger (Konduktor) mit dem Genotyp Aa (=heterozygot) enthält sowohl das Allel A als auch das rezessive Allel a, welches das veränderte Hämoglobin S verursacht. Seine roten Blutkörperchen enthalten HbA und HbS im Verhältnis 1:1. Unter Normalbedingungen zeigen die roten Blutkörperchen keine Veränderungen, die Krankheit kommt nicht zum Ausbruch. Erst unter sehr starkem Sauerstoffmangel verformen sich die roten Blutkörperchen zu sichelförmigen Gebilden, was die Durchblutung der Organe beeinträchtigt.
- Ein Träger des Genotyps aa (=homozygot) stellt nur das veränderte HbS her. Schon unter physiologischem Sauerstoffmangel, wie er z. B. in den Kapillaren Sauerstoff verbrauchender Organe herrscht, kommt es zu einer starken Verformung der roten Blutkörperchen. Sie verlieren ihre Elastizität und verhaken leicht miteinander. Dadurch kommt es zu einem Verschluss der Kapillaren. Unter normalen Bedingungen ist das Hämoglobin in den roten Blutkörperchen stets fein verteilt. Bei abnehmendem pH-Wert und Sauerstoffgehalt des Blutes kommt es beim HbS zu einer Verklumpung der Hämoglobinmoleküle zu stäbchenförmigen, kristallinen Gebilden. Dadurch wird der Erythrozyt sichelförmig verformt und verliert seine Elastizität.
Diagnose
Stammbaumanalyse
Sind die Genotypen der Eltern bekannt, kann mit Hilfe des Hardy-Weinberg-Gesetzes die Wahrscheinlichkeit berechnet werden, mit der die Sichelzellenanämie bei einem Kind auftritt:
Genotyp der Eltern Genotypen der Kinder Phänotyp der Kinder AA x AA AA100 % gesunde Kinder AA x Aa AA50 % Wahrscheinlichkeit, dass ein Kind gesund ist Aa50 % Wahrscheinlichkeit, dass ein Kind zwar gesund ist aber Überträger Aa x Aa AA25 % Wahrscheinlichkeit, dass ein Kind gesund ist Aa50 % Wahrscheinlichkeit, dass ein Kind zwar gesund ist aber Überträger aa25 % Wahrscheinlichkeit, dass ein Kind mit Sichelzellenanämie zur Welt kommt AA x aa Aa100 % alle Kinder sind mit Sicherheit zwar gesund aber Überträger Aa x aa Aa50 % Wahrscheinlichkeit, dass ein Kind zwar gesund ist aber Überträger aa50 % Wahrscheinlichkeit, dass ein Kind mit Sichelzellenanämie zur Welt kommt aa x aa aa100 % Die Kinder haben mit Sicherheit Sichelzellenanämie Gelelektrophorese
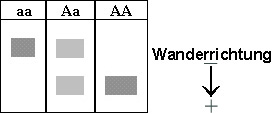
Da erst unter extremem Sauerstoffmangel eine Veränderung der roten Blutkörperchen von Überträgern (Genotyp Aa) auftritt, lässt sich durch Untersuchung der roten Blutkörperchen unter dem Mikroskop der Genotyp AA nicht vom Genotyp Aa unterscheiden. Dagegen lässt sich mit Hilfe der Elektrophorese eindeutig der Genotyp bestimmen: Dazu wird den Probanden Blut entnommen und aufbereitet, bis reines Hämoglobin vorliegt. Dieses wird auf ein Gel aufgetragen. Im elektrischen Feld wandern die beiden Hämoglobinsorten unterschiedlich weit, da HbS aufgrund seiner Mutation langsamer ist als HbA.
Restriktionsanalyse
Zufällig enthält der Abschnitt des β-Globulin-Gens eine Schnittstelle für das Restriktionsenzym MstII mit der Erkennungssequenz 5´-CCTNAGG-3´ (wobei N = irgendeine Base). Durch die Punktmutation wird die Schnittstelle für MstII zerstört. Das bedeutet, dass die Mutation einen Restriktionsfragment-Längenpolymorphismus (RFLP) zur Folge hat.
Vorgehen:
- Isolierung der DNA
- Vervielfältigung durch PCR (Polymerasekettenreaktion)
- Zugabe von MstII
- Fragmentlängenanalyse per Gelelektrophorese wobei mutierte DNA-Fragmente aufgrund ihrer größeren Länge weniger Weg zurücklegen als die DNA-Fragmente gesunder Personen
Verbreitung
Auffallend ist, dass in Gebieten der Malaria das Sichelzellenallel relativ häufig ist. Dies erklärt sich daraus, dass es gegen Malaria eine Resistenz verleiht, sodass die gesunden Überträger (Aa) Träger des Sichelzellenallels in diesen Gebieten einen Evolutionsvorteil (den sogenannten „Heterozygotenvorteil“) gegenüber denen ohne Sichelzellenallel (Genotyp AA) haben, die eher an Malaria sterben, und auch gegenüber den an Sichelzellenanämie Leidenden (Genotyp aa) haben, die vorzeitig an Sichelzellenanämie sterben. In Afrika gibt es beispielsweise Gegenden, in denen fast ein Drittel der Bevölkerung heterozygot für dieses Merkmal ist. In den anderen Weltgegenden kommt das Sichelzellenallel praktisch nicht vor, da hier dieser Selektionsvorteil aufgrund der fehlenden Malaria nicht wirksam ist. In Deutschland werden jährlich etwa 300 Ausbrüche festgestellt. In der schwarzafrikanischen Bevölkerung tritt Sichelzellenanämie mit einer Häufigkeit von 1:250 auf und führte bei homozygoten Trägern unbehandelt oft vor dem 30. Lebensjahr zum Tod.
Malariaresistenz
Der genaue Mechanismus der Malariaresistenz ist zum momentanen Zeitpunkt noch nicht bekannt. Dennoch bestehen einige Theorien, deren Zusammenklang eine Vermutung über die Funktion der Sichelzellenanämie als Selektionsvorteil bei Malaria zulassen.
Zunächst ist zu bemerken, dass die Malariasporozoiten (siehe Hauptartikel Malaria) mit dem Blut - also mit den Erythrozyten transportiert werden. Konstruieren wir also den Fall, eine Person, die Konduktor der Sichelzellanämie ist, würde von einer Mücke gestochen, die den Malariaparasiten trägt. Das Hämoglobin dieses Menschen würde durch Verminderung des Sauerstoffpartialdruckes unter Extrembedingungen zur sichelartigen Verformung der roten Blutzellen führen. Diejenigen Zellen, die vom Malariaparasiten betroffen sind, würden sich auch ohne diese Druckverminderung und schon allein durch den Einfluss der Sporozoiten verformen, von der Milz als krank erkannt und abgebaut werden.
Eine weitere Möglichkeit wäre die direkte Tötung der Parasiten, da diese unter Einfluss des HbS überdurchschnittlich viele Vakuolen produzieren. Außerdem bilden die Sichelzellen vermehrt Sauerstoffradikale. Es entstehen dabei Superoxidanionen und Wasserstoffperoxid, beide Verbindungen sind für die Parasiten giftig.
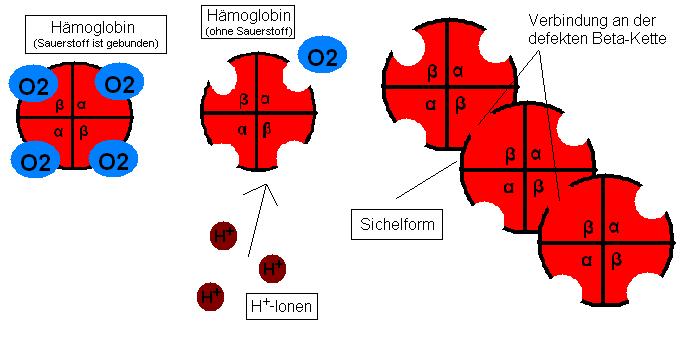
 H+-Ionen begünstigen die Sauerstoffabgabe und die defekten β-Ketten können sich verbinden. Es kommt zur Sichelform.
H+-Ionen begünstigen die Sauerstoffabgabe und die defekten β-Ketten können sich verbinden. Es kommt zur Sichelform.Eine andere Theorie besagt Folgendes: Befallen die Plasmodien, die die Malaria verursachen, Erythrozyten, so setzen die Mikroben nach einiger Zeit Säuren als Abfallprodukte ihres Stoffwechsels frei. Das Hämoglobin gibt nun von H+-Ionen angeregt den Sauerstoff ab. Die Sichelform betrifft aber vor allem die Desoxyform der Erythrozyten. Also werden die befallenen Zellen schnell zu Sichelzellen und diese werden dann in der Milz samt Mikroben abgebaut. Daraus erklärt sich die Resistenz der Träger der Sichelzellenanämie gegenüber Malaria. (siehe Abbildung)
Die letzte Theorie besagt, dass dabei unter der Bildung von Hämoglobinpolymeren Hämin entsteht, das wiederum zur direkten Tötung der Parasiten führt.[2]
Quellen
- ↑ Harrisons Innere Medizin, 15. Auflage, S 1754
- ↑ Skript der Harvard Medical School (englisch)
Weblinks
- Ausführlicher praktischer Leitfaden der Uni Bonn
- Sickle Cell Disease (engl.) Webseite der Harvard-Universität zu den Sichelzellkrankheiten.
- "Video"-Darstellung der Veränderung
- Sickle Cell Anemia (engl.) Ausführliche Seite von U.S. National Library of Medicine und National Institutes of Health

Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.