- Dünnschichtchromatografie
-
 DC eines Tintenfarbstoffgemisches
DC eines TintenfarbstoffgemischesDie Dünnschichtchromatografie (DC, englisch: thin layer chromatography TLC) ist ein physikalisch-chemisches Trennverfahren, das zur Untersuchung der Zusammensetzung von Proben genutzt wird. Besonders vorteilhaft sind: der geringe apparative Aufwand, die Schnelligkeit der Methode, die hohe Trennleistung und der geringe Substanzbedarf. Eingesetzt wird sie zum Beispiel zum raschen Nachweis von Reinheit und Identität oder zur Verfolgung des Reaktionsverlaufes von chemischen Umsetzungen im Labor. Untersucht werden zum Beispiel Probelösungen von Naturstoffen, Medikamenten, Reaktionsmischungen und Reaktionsprodukten im chemischen Labor. Außerdem kann durch geeignete Techniken auch eine Quantifizierung vorgenommen werden. Lösungen von bekannten Vergleichssubstanzen werden dabei oft gleichzeitig mit der eigentlichen Probe untersucht und dienen zur sicheren Identifizierung der chemischen Verbindungen in der Probe. Grundlage der Methode sind Wanderungsprozesse in einer Flüssigkeit, die durch eine Pulverschicht strömt. Dabei zeigen unterschiedliche Moleküle meist unterschiedliches Wanderungsverhalten. Die Methode ist vielfältig einsetzbar und weit verbreitet. Alle Proben, die ausreichend stabil sind und in Lösung gebracht werden können, lassen sich untersuchen. Der Erfolg hängt davon ab, wie groß die Unterschiede beim Wanderungsverhalten der interessierenden Moleküle sind. Farblose Substanzen können durch geeignete Methoden sichtbar gemacht werden. Dazu gehört neben der Fluoreszenzlöschung auch die Reaktion mit Sprüh- oder Tauchreagenzien (Derivatisierung). Eine andere Methode ist die Derivatisierung mit Chromophoren vor der Trennung. Im größeren Maßstab dient die Methode auch zur Reinigung von Substanzen (präparative Dünnschichtchromatografie bzw. auch Dickschichtchromatografie).
Oberbegriffe sind Flüssigchromatografie und Chromatografie.
Inhaltsverzeichnis
Geschichte [1]
N. A. Izmailov und M. S. Shraiber, zwei russische Forscher, führten 1938 eine chromatographische Trennung mit einer horizontalen Dünnschichtplatte durch, auf die sie das Lösemittel auftropften. Doch ihre Methode wurde kaum beachtet. Erst als J. G. Kirchner und seine Mitarbeiter (darunter auch B. Harnischmacher) sich ab 1951 mit ihr befassten, wurde das Interesse anderer an der Methode geweckt. Zum Durchbruch verhalf ihr Egon Stahl, als er die Herstellung von leistungsfähigen Platten beschrieb. Von ihm stammt auch der Name Dünnschichtchromatografie.
Phasen
Bei einer genaueren Analyse dieses Begriffes sollte man von einem Wettkampf zwischen den diversen Probemolekülen mit den Molekülen des Fließmittels (Laufmittels) um die Haftstellen des Adsorbens sprechen. In der Regel handelt es sich dabei um polare Haftstellen (z. B. bei Kieselgel, Normalphasenchromatographie). Sehr interessant ist auch die Verwendung von DC-Material, bei dem Adsorbentien eingesetzt werden, die unpolare Haftstellen aufweisen (Umkehrphasenchromatographie, reversed phase). Die Reihenfolge, in der die verschiedenen Probemoleküle aufgetrennt werden, ist dabei umgekehrt (die polaren Moleküle laufen weiter). Vorteilhaft ist dabei unter anderem, dass auch sehr polare Proben untersucht werden können oder Proben unmittelbar aus wässriger Lösung heraus (die Polarität des Fließmittels ist im allgemeinen ebenfalls höher). Es gibt auch DC-Material mit speziellen chemischen Gruppen im Adsorbens (so genannte chemisch gebundene Phasen) für Spezialanwendungen. Ein Beispiel dafür ist die Chiralplate[2] genannte reversed phase-DC-Platte die mit dem Kupfer-Komplex eines chiralen Derivates[3] der Aminosäure L-Prolin beschichtet ist und die direkte dünnschichtchromatographische Trennung von Enantiomeren nach dem Prinzip der chiralen Ligandenaustauschchromatografie erlaubt[4].
Theoretische Grundlagen
Grundprinzip der Auftrennung
Das Grundprinzip der Chromatografie gilt für alle chromatographischen Methoden und kann wie folgt kurz zusammengefasst werden: Teilchen (Moleküle, Ionen) verteilen sich auf zwei Phasen in einem bestimmten Verhältnis (Gleichgewichtszustand). Entscheidend ist, dass sie individuell sehr rasch von der einen Phase in die andere wandern (auf Grund der Wärmebewegung, der Diffusion und der raschen Austauschprozesse) und wieder zurück (dynamischer Gleichgewichtszustand). Der Zeitanteil, den das individuelle Teilchen in der mobilen bzw. der stationären Phase zubringt, entspricht auch genau dem Anteil der Teilchen dieser Sorte in den beiden Phasen. Diese Verhältnisse gelten auch, wenn die mobile Phase nicht bewegt wird.
Der „Trick“ der Chromatografie besteht einfach darin, die Unterschiede, die es bei diesen Austauschvorgängen zwischen den verschiedenen Teilchensorten gibt, in Geschwindigkeitsunterschiede zu verwandeln. Die Geschwindigkeit ergibt sich aus dem Produkt der Geschwindigkeit der mobilen Phase und dem Zeitanteil, den die Teilchen in der mobilen Phase verbringen. Es wird angenommen, dass die Teilchen in der mobilen Phase (statistisch gesehen) dieselbe Geschwindigkeit haben wie die Laufmittelmoleküle. Sind sie an die stationäre Phase gebunden, ist die Geschwindigkeit gleich Null („stop and go“-Modell). So werden also Verteilungsunterschiede (Verteilung im allgemeinen Sinn) in Geschwindigkeitsunterschiede verwandelt. Oft sind die Verteilungsunterschiede nur gering. Auftrennungen wären so mit anderen Methoden kaum zu erzielen. In dem Augenblick, in dem es sich aber um Geschwindigkeitsunterschiede handelt, ist es nur eine Frage der Länge der Wegstrecke, bis es zu einer ausreichenden Auftrennung gekommen ist.
Zonenverbreiterung
Bestimmte statistische Umstände (verschiedene Arten der Diffusion) wirken einer guten Auftrennung entgegen. Entscheidend ist der rasche Wechsel der Teilchen zwischen Phase 1 und Phase 2. Daher ist es auch günstig, möglichst geringe Laufstrecken und feine, einheitliche Korngrößen des Schichtmaterials zu haben. Das ist auch günstig, um die Unterschiede der gesamten Wegstrecke, die für eine bestimmte Teilchenart auftreten, gering zu halten („Zickzack-Weg“). Zu geringe und zu hohe Geschwindigkeiten der mobilen Phase wirken sich negativ aus. Eine zu geringe Geschwindigkeit begünstigt eine Vergrößerung der Zonen, in denen sich die Probemoleküle aufhalten. Je mehr Zeit zur Verfügung steht, desto größer ist die Rolle, die Diffusionsprozesse innerhalb der mobilen Phase spielen. Ist die Geschwindigkeit zu hoch, kommt es seltener zu einem Wechsel der Teilchen zwischen den Phasen 1 und 2. Das führt zu einer größeren statistischen Streuung und ist ebenfalls unerwünscht. Bei allen chromatografischen Methoden gibt es entsprechend eine optimale Geschwindigkeit der mobilen Phase (siehe auch Van-Deemter-Gleichung). Je feiner die Korngrößen sind (bzw. die Dimensionen), desto größer kann sie werden. Das ist auch ein ökonomischer Vorteil.
Bei der DC ist die erwünschte räumliche Auftrennung zwischen den verschiedenen Probekomponenten der gesamten Laufstrecke proportional (Abstand Startlinie – Laufmittelfront). Die Vergrößerung der einzelnen Zonen auf Grund statistischer Effekte ist geringer (nicht der Laufstrecke proportional sondern der Wurzel der Laufstrecke). Daher ist es bei schwierigen Trennungen sinnvoll, größere DC-Folien und Laufstrecken zu verwenden.
Durchführung
Auftragen
Die zu untersuchende Substanz wird in einem geeigneten Lösungsmittel gelöst und mit Hilfe einer Kapillare punkt- oder strichförmig aufgetragen. Die zu trennende Substanzmenge beträgt ca. 5-20 Mikrogramm. Für eine besonders gleichmäßige und auch quantitativ reproduzierbare Auftragung stehen auch Maschinen zur Verfügung, welche die Lösung mit Hilfe von Druckluft oder Stickstoff aufsprühen. Die stationäre Phase (Trennschicht) besteht aus einer dünnen Schicht eines sehr feinkörnigem Materials (z. B. Kieselgel, Kieselgur, Aluminiumoxid, Cellulose). Diese Trennschicht ist sehr gleichmäßig auf eine Trägerfolie oder Trägerplatte aus Kunststoff, Aluminiumblech oder Glas aufgetragen und kommerziell in unterschiedlichen Schichtdicken erhältlich. Auf die DC-Folie wird auf der Startlinie, bei der 2-dimensionalen DC in einer Ecke, das zu untersuchende Gemisch aufgetragen.
Daneben werden auf der Startlinie in vielen Fällen auch Lösungen von reinen Vergleichssubstanzen oder Vergleichsmischungen aufgetragen. Es ist entscheidend, die Auftragzonen möglichst eng zu halten (wenige Millimeter). Kommerziell erhältlich sind auch DC-Folien mit so genannten Konzentrationszonen. Hier wird eine Zone mit besonders geringer Adsorption vorgeschaltet. Die Probeflecken werden dadurch nach Beginn der Chromatografie in der Richtung der Laufstrecke zusammengestaucht und so sind besonders enge Auftragzonen erzielbar.
Nach dem Auftragen muss die Platte getrocknet werden, da restliches Lösungsmittel das Ergebnis verändern kann.
Für spezielle Anwendungen kann auch das „Waschen“ der Platte vor dem Auftragen der Probe oder auch das Trocknen im Exsikkator oder Trockenschrank bei erhöhter Temperatur notwendig sein. Gewaschen werden die Platten durch Einstellen in eine Chromatographiekammer mit dem entsprechenden Lösungsmittel bis die Laufmittelfront die Oberkante der Platte erreicht hat.
Auftrennung
Nach dem Auftragen wird die Platte senkrecht in eine Chromatographiekammer mit einem geeigneten Fließmittel (mobile Phase) eingestellt. Um eine Beeinflussung der Ergebnisse durch das Verdampfen des Fließmittels zu verhindern, führt man die Trennung in einer mit dem Fließmittel gesättigten Atmosphäre in einem geschlossenen Gefäß durch. Zur besseren Sättigung des Dampfraumes mit Laufmittel kann ein Filterpapier eingelegt werden. Die Sättigung verhindert Verdampfen des Laufmittels von der Platte und damit eine Änderung der Zusammensetzung auf der Platte. Als Laufmittel werden in der Normalphasen-DC unpolaren organische Lösungsmittel in der Regel als Gemisch mit mäßig polaren Lösungsmitteln genutzt (z. B. Petrolether und Essigsäureethylester); in der Umkehrphasen-DC dagegen polare Laufmittel (z.B. Acetonitril). Über das Mischungsverhältnis kann man somit die Polarität des Fließmittels steuern. Das Fließmittel saugt sich nun über Kapillarkräfte in die stationäre Phase nach oben. Sobald die Flüssigkeit die Startlinie erreicht hat, lösen sich die Substanzen in ihr. Die Moleküle sind nun den Anziehungskräften der stationären Phase einerseits und den Anziehungskräften der mobilen Phase andererseits ausgesetzt. Je nach Kräfteverhältnis bleibt ein Teilchen eher am Startpunkt oder es wandert mit der mobilen Phase nach oben. Im Allgemeinen gilt: Je unpolarer das Fließmittel ist, desto weniger wandern polare Substanzen und umgekehrt. Es gilt aber auch, dass polare Substanzen mit polaren stationären Phasen stärker wechselwirken. Die Lösungsmittelpolarität ist dabei analog wie in der Säulenchromatografie. Mit der DC-Chromatografie lässt sich auf einer 15cm langen Laufstrecke eine Trennleistung von ca. 400-3000 theoretischen Böden erzielen.
Durch unterschiedliche Wechselwirkungen der Komponenten des aufgetragenen Gemischs mit der stationären und der mobilen Phase trennt sich die Mischung auf. Es lassen sich sogar Spiegelbildisomere (= Enantiomere) trennen, wenn man eine mit einem chiralen Selektor modifizierte stationäre Phase verwendet. Derartige gebrauchsfertige Platten sind unter dem Handelsnamen Chiralplate® (D) erhältlich.
Die Kräfte und somit das Wanderverhalten eines Teilchens hängen sowohl von der Art des Schichtmaterials und des Fließmittels, als auch von der Art des Teilchens ab. In den meisten Fällen lassen sich Schichtmaterialien und Fließmittelgemische so kombinieren, dass die verschiedenen Teilchensorten eines Gemisches verschieden weit wandern, sodass sie sich von einander trennen lassen.
Bei der zweidimensionalen DC wird nach der ersten Entwicklung das Laufmittel abgedampft, die Platte um 90° gedreht und, in der Regel in einem anderen Laufmittel, eine zweite Entwicklung durchgeführt. Dadurch kann eine bessere Auftrennung bei Multikomponentengemischen erreicht werden. Die Identifizierung ist aber aufwendiger, da keine Referenzsubstanzen mitlaufen können.
Auswertung
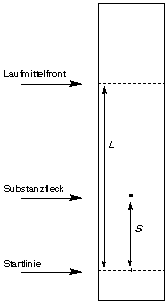 Schematische Darstellung einer DC-Platte
Schematische Darstellung einer DC-PlatteDie getrennten Substanzen werden durch Besprühen mit Reagenzien-Lösungen sichtbar gemacht (detektiert). Alternativ ist bei der Anwendung auch Tauchung in Lösungen möglich. Eine andere Möglichkeit ist das Derivatisieren vor der Chromatographie mit Chromophoren und das Betrachten unter UV-Licht.
Bei der Verwendung von Sprüh- oder Tauchreagenzien kommen Farbreaktionen zum Einsatz, die empfindlich und spezifisch genug sind, um zum Nachweis bestimmter funktioneller Gruppen verwendet zu werden. Über die Auswahl der Farbreaktion lässt sich der Informationsgehalt bei der DC wesentlich erhöhen. Alternativ werden Reaktionen eingesetzt, die allgemein wirksam sind (zum Beispiel Oxidation mit Hilfe von Salpetersäurelösungen oder Ioddampf). Bei einer Reihe von Farbreaktionen ist es erforderlich, die Folie nach dem Besprühen bzw. der Tauchung zu erhitzen.
Viele Schichtmaterialien enthalten Zusätze, die im UV-Licht fluoreszieren und an denjenigen Stellen dunkle Fluoreszenzquenchung (Fluoreszenzlöschung) zeigen, an denen sich die getrennten Stoffe befinden. Diese Fluoreszenzfarbstoffe dürfen während der chromatografischen Trennung nicht wandern. Gebräuchlich sind vor allem Mangan aktiviertes Zinksilikat (mit UV-Licht der Wellenlänge 254 nm bestrahlt) und Calciumwolframat (mit UV-Licht der Wellenlänge 366 nm bestrahlt). Tatsächlich handelt es sich bei der Methode nicht um eine Fluoreszenzlöschung im engeren Sinne. Probemoleküle werden sichtbar, wenn sie im Bereich von 254 nm oder 366 nm UV-Licht absorbieren. Es gelangt dann weniger UV-Licht zu den Fluoreszenzfarbstoffmolekülen (dunkle Flecken auf grün bzw. blau leuchtendem Hintergrund zu sehen). Dazu müssen genügend viele funktionelle Gruppen bzw. genügend große Systeme mit konjugierten Doppelbindungen vorhanden sein. Gesättigte Kohlenwasserstoffe und viele Aminosäuren sind daher mit dieser Methode nicht nachzuweisen, aromatische Verbindungen z. B. sehr leicht bei 254 nm.
Auch die Eigenfluoreszenz bestimmter Stoffe oder andere Eigenschaften wie Radioaktivität können zur Detektion herangezogen werden.
Eine weitere, sehr einfache Methode besteht in der Bedampfung mit molekularem Iod. Dazu genügt es, in ein Glasgefäß ein paar Iod-Kristalle zu legen. Sie sublimieren, das heißt sie verdampfen direkt bei Raumtemperatur, bilden einen violetten Dampf von Diiodmolekülen. Durch Einlegen einer DC-Folie in einen solchen Trog werden über Diffusion und Reaktion mit den Molekülen der Substanzflecken binnen kurzer Zeit lockere Komplexverbindungen gebildet (lila oder braun). Vorteil bzw. Nachteil der Methode: die Iod-Verbindungen zerfallen relativ rasch.
In der Biochemie ist eine saure Ninhydrin-Lösung ein häufiges Sprühreagenz, um Aminosäuren zu detektieren. Hierbei wird das Ninhydrin über die Schiffsche Base und durch eine Decarboxylierung sowie Hydrolyse zu Ruhemans Violett. Durch Auftragen von Referenzproben, welche unter gleichen Bedingungen gleich weit wandern wie entsprechende Probekomponenten, kann man das qualitative Auftreten von Stoffen nachweisen. Hierzu wird die Lage der verschiedenen Punkte mit der Lage der Referenzproben verglichen.
Um verschiedene DC vergleichen zu können werden die so genannten Rf-Werte (Retentionsfaktor, Rückhaltefaktor, ratio of fronts) berechnet. Es handelt sich dabei um das Verhältnis Wanderungsstrecke des Substanzfleckes (S) zur Wanderungsstrecke des Lösemittels (L):
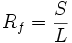 . Die Rf-Werte sind bei gleichem Plattenmaterial und gleichem Laufmittel Stoffkonstanten.
. Die Rf-Werte sind bei gleichem Plattenmaterial und gleichem Laufmittel Stoffkonstanten.Sinnvoll und gebräuchlich ist eine Kombination von Methoden: zuerst Betrachtung unter der UV-Lampe, anschließend chemische Reaktionsmethoden.
Die quantitative Auswertung kann mit einem Densitometer ausgeführt werden.
Präparative DC
Die DC kann auch in Form der PLC (preparative layer chromatography) präparativ genutzt werden. Hierzu werden auf dickere stationäre Phasen größere Mengen der zu trennenden Substanzmischung strichförmig aufgetragen. Nach dem Trennlauf bilden sich völlig voneinander getrennte Striche in verschiedenen Höhen, die dann z. B. durch mechanisches Abkratzen in verschiedene Gefäße verteilt und separat eluiert werden.
Für bestimmte, besonders empfindliche Analyseverfahren (Massenspektrometrie, Infrarotspektroskopie) lassen sich auch die üblichen dünnen (analytischen) DC-Folien präparativ nutzen.
Empfehlenswert ist auch die so genannte zirkulare Dünnschichtchromatografie. Dabei werden kreisförmige Glasscheiben beschichtet (eine Kreiszone). Das Schichtmaterial enthält einen Fluoreszenzfarbstoff (der bei UV-Licht der Wellenlänge 254 nm oder 366 nm fluoresziert). Die Probelösung wird mit Hilfe einer Pumpe zum inneren Rand der Schicht geleitet, vorher und nachher das entsprechende Fließmittel.
Die Scheibe sitzt dabei auf einem Rotor auf und wird mit Hilfe eines Elektromotors in rasche und gleichmäßig kontrollierte Rotation versetzt. Zur Beobachtung wird der Trennvorgang unter Bestrahlung mit einer UV-Lampe kontrolliert. Am Anfang des Trennprozesses befindet sich die Probe in einer wenige Millimeter starken Kreiszone am inneren Rand der Scheibe. Im Lauf der Zeit zerlegt sich die Probe in eine Reihe von Ringen, die nach außen wandern.
Wenn eine Probenkomponente am äußeren Rand der Schicht angelangt ist, wird sie zusammen mit dem Laufmittel in einen Kunststoffreifen zentrifugiert, der die Führung der Scheibe umgibt. Die Achse der Scheibe ist während der Chromatografie schräg gestellt, das „Eluat“ rinnt am unteren Ende des Kunststoffreifens zusammen und über eine Öffnung und einen Schlauch in entsprechende Sammelbehälter.
Die Methode hat eine Reihe von Vorteilen (gegenüber normaler DC und gegenüber der üblichen präparativen Säulenchromatografie):
- Es können größere Mengen aufgetrennt werden als mit normaler Dickschichtchromatografie (bei geringerem Zeitaufwand).
- Die Oberfläche der Schicht kann gut zugeschliffen werden (einheitliche Schichtdicke).
- Das kostspielige Schichtmaterial wird gut für den Trennprozess ausgenützt („Geometrie“ ist ideal).
- Es ist einfach möglich, während der Chromatografie die Polarität des Laufmittels (Fließmittels) zu erhöhen. Davon wird gerne Gebrauch gemacht. Damit lassen sich auch Mischungen zerlegen, die Substanzgruppen enthalten, die sich voneinander stark in der Polarität unterscheiden.
- Am Schluss ist es einfach möglich, die Schicht durch längeres Auswaschen mit einer polaren Laufmittellösung wieder zu reinigen. Der Reinigungsprozess lässt sich gut verfolgen (UV-Lampe). Im Gegensatz zur Regeneration bei Säulenchromatografie-Methoden kann diese „Waschflüssigkeit“ leicht entfernt werden: Abdampfen lassen, trocknen und aktivieren in einem elektrischen Ofen (ca. 50 bis 60 Grad Celsius).
- Der Fortgang der Chromatografie kann ständig verfolgt werden. Die Aufsammlung der Probekomponenten wird dadurch erleichtert (das „Schneiden“ der Fraktionen). Der Trennprozess lässt sich adaptieren, entsprechend der laufenden Beobachtung.
- Die Aufgabe der Probe ist einfacher zu gestalten als bei der normalen präparativen DC. Durch Verwendung eines Lösungsmittels mit niedriger Polarität lässt sich eine besonders enge („scharfe“) Startzone erreichen.
Nachteile:
- Bei luft- und UV-licht-empfindlichen Verbindungen kann es Probleme geben. Unter Umständen kann auch unter Schutzgas gearbeitet werden.
- Die Methode kann nicht angewendet werden, wenn die Substanz UV-Licht nicht absorbiert.
- Die Anschaffungskosten des Gerätes sind im Vergleich zu DC-Kammern relativ hoch. Die Kunststoffteile bestehen aus Teflon.
Vorteile und Nachteile der Dünnschichtchromatografie
Im Gegensatz zu den leistungsfähigeren Chromatografie-Verfahren wie Gaschromatografie und Hochleistungsflüssigkeitschromatographie kommt die DC mit geringem apparativen Aufwand aus und stellt sich als schnelles, vielseitiges und preiswertes Analyseverfahren dar.
Die Gaschromatografie lässt sich nur bei Proben anwenden, die unzersetzt verdampfbar sind. Bei der Flüssigchromatografie gibt es wenig Einschränkungen. Fast immer lässt sich ein Weg finden, eine Probe aufzulösen. Im Vergleich zu den säulenchromatografischen Methoden besteht der Vorteil, dass Proben, die Gruppen von Komponenten enthalten, die sich jeweils stark in der Polarität unterscheiden, leichter zu erfassen sind. Laufmittelwechsel ist nicht so leicht möglich wie bei der Säulenchromatografie. Es ist aber möglich, zunächst in einem Laufmittel zu entwickeln und nach Zwischentrocknung in einem anderen (das sich in der Polarität stark unterscheidet).
Nachteilig ist bei der analytischen Anwendung der DC, dass es schwerer ist, eine quantitative Analyse durchzuführen. Bei bestimmten Aufgabenstellungen genügt es allerdings schon, die Mengenverhältnisse abzuschätzen (Fortschritt einer chemischen Reaktion).
Beispielexperiment: Dünnschichtchromatografie von Blattfarbstoffextrakt
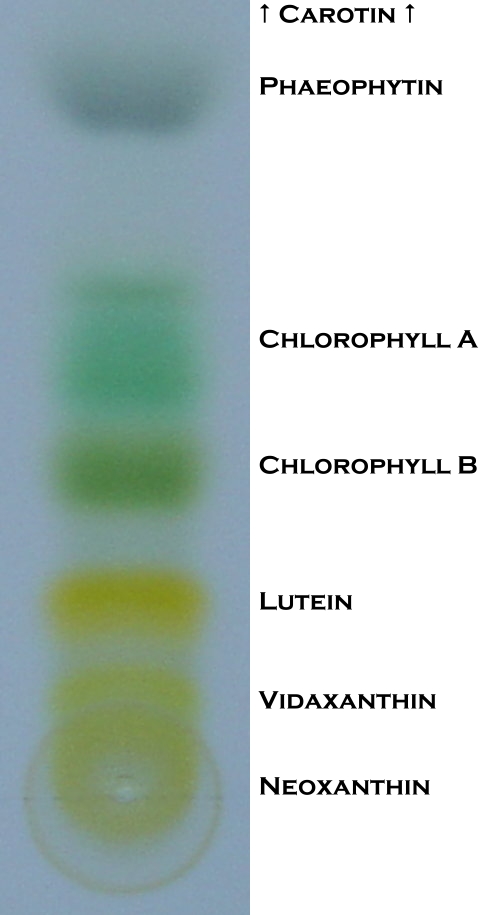
Durch dünnschichtchromatografische Untersuchungen von Blattfarbstoffextrakt kann man erkennen, aus welchen Farb-Komponenten er besteht: Carotinen, Chlorophyllen und Xanthophyllen.
Versuchsmaterial
- mit einem adsorbierenden Pulver (z. B. Kieselgel) fertig beschichtete Aluminium- oder Kunststofffolie(n) bzw. Glasplatte(n), Blattfarbstoffextrakt (Herstellung s. unten), Fließmittel (am besten geeignet: Benzin-Isopropanolgemisch, Mischverhältnis 10:1), Kapillarröhrchen (sehr klein, oder alternativ: Mikro-Pipette oder dünner Pinsel), Apfelmus- oder anderes Glas
- für den Blattfarbstoffextrakt: frische, grüne Blätter, z. B. von Spinat oder Salat (im Winter auch Wintersalat), Aceton (20 ml), Calciumcarbonat (Spatelspitze), (evtl. gereinigter) Meeres- bzw. Seesand (Spatelspitze), Mörser, Pistill, Erlenmeyer-Kolben, Trichter, Faltenfilter, Aluminiumfolie (nicht zwingend nötig: Haartrockner/Fön, Bleistift, Lineal)
Herstellung des Chlorophyllextraktes/ Isolierung der Blattpigmente
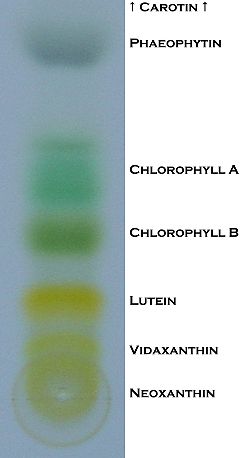 Ergebnis
ErgebnisCa. 5 Gramm Blattmaterial werden unter Verwerfung der starken Blattrippen sowie der Stiele in kleine Stücke zerrissen und in den Mörser gelegt. Zum zerkleinerten Blattmaterial wird das Calciumcarbonat hinzugegeben, dies dient der Neutralisation des sauren Zellsaftes. Daraufhin kommt Seesand in den Mörser, um die Blattstücke aufschließen zu können. Nach der Zugabe von Aceton wird das Material im Mörser einige Minuten zerrieben. Wenn eine kräftig dunkelgrün gefärbte Lösung entstanden ist, wird sie in einen Erlenmeyer-Kolben abfiltriert.
Achtung: Die entstandene Lösung ist sauerstoff-, licht- und wärmeempfindlich, muss deshalb in einem mit Aluminiumfolie umwickelten Gefäß im Kühlschrank aufbewahrt werden. Nun kann das Extrakt verwendet werden (nicht trinken, lösemittelhaltig!).
Versuchsdurchführung
Der gewonnene Blattfarbstoffextrakt wird mit einem Kapillarröhrchen, einer Mikro-Pipette oder einem sehr feinen Pinsel auf eine hochkant gelegte, beschichtete (raue) Seite der Folie (bzw. Platte) sparsam aufgetragen. Dies kann, je nach Methode, punktförmig (Abstand von unten: etwa 1,5 cm), oder linienförmig (etwa 1,5 cm parallel zur unteren Kante) geschehen. Die Auftragslinie kann auch mit einem feinen Bleistift gekennzeichnet werden. Dabei sollte die Kieselgelschicht möglichst wenig verletzt werden. Nachdem der Extrakt auf der Folie getrocknet ist (dies kann durch einen Haartrockner beschleunigt werden – mit kaltem Luftstrom), kann dieser Auftrage-Vorgang wiederholt werden. Dies soll etwa 10 bis 15 mal passieren. Ist auf der Folie dann der grüne Punkt (bzw. die Linie) wieder getrocknet, wird die Folie in das mit ein wenig Fließmittel gefüllte Apfelmusglas gestellt, danach wird es geschlossen. Das Glas sollte jetzt nicht mehr oder nur sehr vorsichtig berührt werden, da nun der Fließvorgang beginnt. Sobald die Flüssigkeit bis zum Ende des Blattes gestiegen ist (oder schon früher, je nach gewünschtem Ergebnis) wird es aus dem Glas genommen und auf einigen Blatt Papier getrocknet.
Die Farbenpracht ist allerdings nicht von langer Dauer: nach dem Trocknen sind die Gelb-Anteile bereits verblichen. Nach einiger Zeit verbleiben nur noch unterschiedliche Grüntöne auf der Folie.
Hinweis: Je länger der Farbstoffextrakt vor dem Versuch stand, umso größer ist der Grau-Anteil auf der beschichteten Folie. Dies liegt daran, dass der graue Anteil aus Abbaustoffen der Chlorophylle besteht, die erst nach der Extraktherstellung und unter Sauerstoff-, Wärme- oder Lichtzufuhr gebildet werden (Phäophytin). Deshalb ist es ratsam, die Lösung so aufzubewahren wie oben beschrieben.
Erklärung des Fließvorgangs
Das Fließmittel wird nach oben hin von der Folienschicht aufgenommen. Dabei trägt es den Blattfarbstoffextrakt mit sich. Die verschiedenen Adsorptionsfähigkeiten der einzelnen Bestandteile des Blattes sorgen dafür, dass sich das Gemisch in seine Bestandteile aufgliedert. Dank dieser Methode kann man feststellen, aus welchen Komponenten die Blattfarbstoffe bestehen (siehe linke Abbildung).
Galerie: Der Versuch in 7 Bildern

Erster Schritt

Zweiter Schritt

Dritter Schritt

Vierter Schritt

Fünfter Schritt

Sechster Schritt

Siebter Schritt
Diese Abbildungen zeigen die Trennung der einzelnen Farbstoffe in sieben Schritten. Das Carotin verteilt sich weit nach oben und ist deshalb nur bis Abb. 2 sichtbar.
Einzelnachweise
- ↑ Joseph C. Touchstone: Practice of Thin Layer Chromatography, WILEY, 3rd edition, 1992, S. 3–4
- ↑ Spezialschicht für die DC-Enantiomerentrennung. eingesehen am 27. März 2009.
- ↑ Kurt Günther, Jürgen Martens und Maren Schickedanz: Dünnschichtchromatographische Enantiomerentrennung mittels Ligandenaustausch, Angewandte Chemie 96 (1984) 514-515; Angewandte Chemie – International Edition English 23 (1984) 506.
- ↑ (a) Kurt Günther: Thin-layer chromatographic enantiomeric resolution via ligand exchange, Journal of Chromatography 448 (1988) 11-30.(b) Kurt Günther, Maren Schickedanz und Jürgen Martens : Thin-Layer Chromatographic Enantiomeric Resolution, Naturwissenschaften 72 (1985) 149-150.
Weblinks







Wikimedia Foundation.