- Fliessverhalten
-
Physikalische Größe Name kinematische Viskosität Größenart kinematische Viskosität Formelzeichen der Größe ν Größen- und
Einheiten-
systemEinheit Dimension SI m2·s−1 L2·T −1 Physikalische Größe Name dynamische Viskosität Größenart dynamische Viskosität Formelzeichen der Größe η Größen- und
Einheiten-
systemEinheit Dimension SI N·s·m−2 = kg·m−1·s−1 M·L−1·T−1 Anmerkungen auch in Pa·s bzw. mPa·s angegeben Die Viskosität ist ein Maß für die Zähflüssigkeit eines Fluids. Der Kehrwert der Viskosität ist die Fluidität, ein Maß für die Fließfähigkeit eines Fluids. Je größer die Viskosität, desto dickflüssiger (weniger fließfähig) ist das Fluid; je niedriger die Viskosität, desto dünnflüssiger (fließfähiger) ist es.
Normalerweise wird mit dem Begriff Viskosität die Viskosität in Scherung verbunden, es ist allerdings auch möglich die Viskosität in Dehnung zu messen, siehe dazu die Seite Dehnviskosität.
Teilchen zäher Flüssigkeiten sind stärker aneinander gebunden und somit unbeweglicher; man spricht daher auch von der inneren Reibung. Sie resultiert nicht nur aus den Anziehungskräften zwischen den Teilchen des Fluids (Kohäsion).
Bei Feststoffen verwendet man stattdessen die Begriffe der Duktilität, Sprödigkeit und Plastizität. Gelegentlich wird Zähigkeit als Synonym für Viskosität verwendet.
Der Begriff Viskosität geht auf den typisch zähflüssigen Saft der Beeren in der Pflanzengattung Misteln (Viscum) zurück. Aus diesen Misteln wurde der Vogelleim gewonnen, „viskos“ bedeutet also grob „zäh wie Vogelleim“.
Inhaltsverzeichnis
Fließverhalten von Flüssigkeiten
Den Effekt innerer Reibung kann man sich vereinfacht durch die Bewegung zweier übereinander liegender, verzahnter Molekülschichten vorstellen (siehe Abb. Punkt 1). Beim Fließen gleiten die Moleküle aneinander vorbei, und um die Verzahnung zu überwinden, benötigt man eine gewisse Kraft. Den Zusammenhang zwischen dieser Kraft und den Eigenschaften des vorliegenden Fluids definiert die Viskosität. Erkennbar wird dieser Zusammenhang besonders gut an der homologen Reihe der Alkane (kettenförmige Kohlenwasserstoffe), hier steigt die Viskosität mit der Kettenlänge und damit den zunehmenden intermolekular wirkenden Van-der-Waals-Kräften kontinuierlich an. Bei den mittleren Alkanen (ab Nonan, neun C-Atome) hat sie bereits einen Wert ähnlich dem von Wasser.
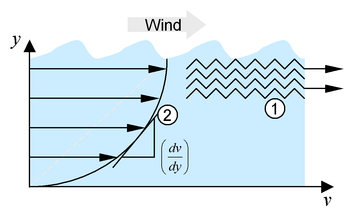 Modellvorstellung zur Viskosität, links der Geschwindigkeitsgradient (2) und rechts eine Veranschaulichung für die verzahnten Molekülschichten (1), weitere Erläuterungen siehe Text
Modellvorstellung zur Viskosität, links der Geschwindigkeitsgradient (2) und rechts eine Veranschaulichung für die verzahnten Molekülschichten (1), weitere Erläuterungen siehe TextSehr gut veranschaulichen kann man sich die Viskosität auch an folgendem Beispiel: gleitet Wind über das Wasser eines Ozeans, erzeugt dies eine Bewegung der Wasserschicht an der Oberfläche. Je tiefer man nun taucht, desto ruhiger wird das Wasser, bis man einen Punkt erreicht, wo keine Strömung herrscht. Die einzelnen Flüssigkeitsschichten bewegen sich mit unterschiedlicher Geschwindigkeit (Korkenzieherströmung), es entsteht ein Geschwindigkeitsgradient (siehe Abb. Punkt 2):
Weht kein Wind mehr, bricht die Strömung zusammen, das Wasser ruht auch wieder an der Oberfläche. Dass die Flüssigkeit auch in tieferen Schichten trotz Wind an der Oberfläche praktisch ruht, ist Folge der inneren Reibung in der Flüssigkeit.
Die dynamische Viskosität der meisten Flüssigkeiten nimmt mit steigender Temperatur ab und kann oft mit der Arrhenius-Andrade-Beziehung beschrieben werden:
Wobei η0 eine Materialkonstante und EA die Aktivierungsenergie (auch Platzwechselenergie), R die allgemeine Gaskonstante und T die absolute Temperatur sind. Bei Flüssigkeiten in der Nähe der Glasübergangstemperatur (bis ca. 100 K darüber) gilt meist die WLF-Beziehung, da das freie Volumen sehr gering ist und damit diese Größe dominiert, die in der Nähe der Glasübergangstemperatur eine sehr viel stärkere Temperaturabhängigkeit hat, als die Kettenbeweglichkeit, die hinter der Arrhenius-Andrade-Beziehung steht.
Definition der Viskosität
Man stelle sich zwei im Abstand x angeordnete Platten der Fläche A vor. Zwischen diesen Platten befindet sich eine Flüssigkeit, die an beiden Platten haftet. In unserer Vorstellung soll der Raum mit der Flüssigkeit in Schichten unterteilt sein. Wird nun Platte 2 mit der Geschwindigkeit v bewegt, so bewegt sich die Schicht in unmittelbarer Nachbarschaft zu Platte 2 auf Grund der Haftung ebenfalls mit der Geschwindigkeit v. Da Platte 1 ruht, ruht auch ihre Nachbarschicht. Die innenliegenden Flüssigkeitsschichten gleiten mit unterschiedlichen Geschwindigkeiten aneinander vorbei. Die Geschwindigkeit nimmt von der ruhenden Platte zur bewegten zu. Im einfachsten Fall besteht eine lineare Abhängigkeit (siehe Abbildung). Von der obersten, an der Platte haftenden Schicht geht eine Tangentialkraft auf die darunterliegende Schicht aus. Diese bewegt sich folglich mit der Geschwindigkeit v1. Diese Schicht wirkt wiederum auf die darunterliegende Schicht und bewegt sie mit der Geschwindigkeit v2.
Im Experiment lässt sich zeigen, dass die Kraft F, die nötig ist, um Platte 2 zu bewegen, proportional zu ihrer Fläche A, ihrer Geschwindigkeit v und antiproportional zu dem Abstand der Platten x ist:
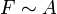 und
und 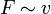 und
und 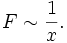
Hieraus ergibt sich
und als Gleichung
Die Proportionalitätskonstante η ist die dynamische Viskosität. Häufig wird sie auch nur als Viskosität bezeichnet. Ein Stoff hat also die Viskosität 1 Ns/m², wenn bei einer Größe der Platten von 1 m² und einem Plattenabstand von 1 m eine Kraft von 1 N benötigt wird, um die Platten mit einer Geschwindigkeit von 1 m/s gegeneinander zu verschieben.
Für die physikalische Einheit gilt:
![1{\rm N}=[\eta] \cdot\left(\frac{{\rm m}^2\,{\rm m}}{{\rm m}\,{\rm s}}\right) \Rightarrow [\eta] = \frac{{\rm N}\,{\rm s}}{{\rm m}^2}.](/pictures/dewiki/97/a52e7d4f20fe0b2dcff8ac2999946d4d.png)
Ist η unabhängig von der Geschwindigkeit v, so wird die Flüssigkeit als Newtonsche Flüssigkeit bezeichnet. Für diese Flüssigkeiten stellt sich das in Abbildung 2 gezeigte, lineare Geschwindigkeitsprofil ein. Ist η von v abhängig, so bezeichnet man die Flüssigkeit als nicht-newtonsch.
Newtonsche Flüssigkeiten
Im Folgenden wird der vereinfachte Zusammenhang gemäß dem newtonschen Viskositätsgesetz dargestellt, es wird dabei stets laminare Strömung sowie Temperatur- und Druckunabhängigkeit der Flüssigkeitseigenschaften angenommen. Außerdem unterstellte Newton eine lineare Abhängigkeit des oben erläuterten Geschwindigkeitsgradienten, der auch Schergeschwindigkeit
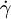 (manchmal auch mit D oder G bezeichnet) genannt wird:
(manchmal auch mit D oder G bezeichnet) genannt wird: Schubspannungs-Schergeschwindigkeits-Diagramm:
Schubspannungs-Schergeschwindigkeits-Diagramm:
1: dilatantes Fluid
2: Newtonsche Fluid
3: Scherverdünnendes (pseudoplastisches) Fluid
4: Bingham-plastisches Fluid
5: Casson-plastisches FluidVerknüpft man dies mit der Schubspannung, erhält man folgenden Zusammenhang für die dynamische Viskosität:
Die Schubspannung τ ergibt sich aus der die Strömung bewirkenden Kraft bezogen auf die betroffene Angriffsfläche, die sich mit maximaler Geschwindigkeit bewegt. η wird bei newtonschen Flüssigkeiten als Konstante angesehen. Darüber hinaus wird das Verhältnis zwischen der dynamischen Viskosität η und der Dichte ρ definiert als kinematische Viskosität:
Nicht-Newtonsche Flüssigkeiten
Viele Substanzen folgen diesem Gesetz jedoch nicht, sondern zeigen ein zeit- oder schergeschwindigkeitsabhängiges Verhalten. Dabei unterscheidet man verschiedene Arten der Abweichung:
- Strukturviskosität / Dilatanz, dabei ist die Viskosität η keine Konstante, sondern ändert sich mit dem Schergefälle
- Thixotropie / Rheopexie, hierbei zeigen sich zeitabhängige Strukturveränderungen, so dass je nach Zeitdauer seit der letzten Fließbewegung andere Viskositätswerte zu finden sind
- Fließgrenze, es muss erst eine gewisse Mindestschubspannung vorhanden sein, um ein Fließen zu erreichen (plastisches Fließen). Diese Art Fluid wird auch als Bingham-Fluid bezeichnet.
Derartige Fluide bezeichnet man als nichtnewtonsche Fluide.
Im allgemeinen Fall muss das Schergefälle
aus dem Scherwinkel in der Flüssigkeit berechnet werden und nicht über den Geschwindigkeitsgradienten.SI-Einheit
Die SI-Einheit der
- dynamischen Viskosität:
![[\eta] = \frac{\rm kg}{\rm m \cdot s} = {\rm Pa} \cdot {\rm s} = \frac{\rm Ns}{\rm m^2}](/pictures/dewiki/54/6e794c798838b1cf3b53bb18de423637.png)
- kinematischen Viskosität:
![[\nu] = \frac{\rm m^2}{\rm s}](/pictures/dewiki/48/02bf4fd293d056d8c0ebc96c381aa866.png)
Im CGS-System wird die dynamische Viskosität in Poise (P) gemessen, wobei 1 Ns/m2 = 1 Pa·s = 10 Poise und 1 Centipoise = 1 cP = 10-3 kg/ms, und die kinematische Viskosität in Stokes (St), 1 St = 10-4 m2/s.Typische Viskositätswerte
 Viskosität verschiedener Flüssigkeiten in Abhängigkeit von der Temperatur
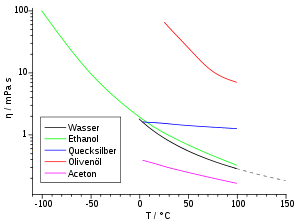
Viskosität verschiedener Flüssigkeiten in Abhängigkeit von der Temperatur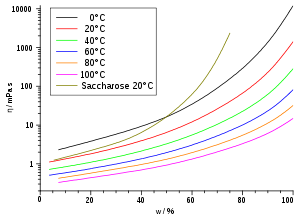 Viskosität von gelöstem Glycerol in Abhängigkeit vom Massenanteil
Viskosität von gelöstem Glycerol in Abhängigkeit vom Massenanteil-
Substanz η in mPa·s 1) Wasser (5 °C) 1,52 Wasser (20 °C) 1,00 Wasser (25 °C) 0,891 Petroleum 0,65 Pentan (25 °C) 0,224 Diethylether 0,240 Hexan 0,320 Heptan 0,410 Octan 0,538 Nonan 0,711 Chloroform 0,56 Decan 0,920 Ethanol 1,19 Essigsäure (80%-ige bei 25°C) 2,31 Benzol (25 °C) 0,601 Glycerin (rein) 1480 Lack ~ 102 Paraffinöl 102 bis 106 Polymerschmelzen 103 bis 1013 2) Bitumen ~ 1011 Asphalt ~ 105 Quecksilber 1,55 Glas (Verarbeitungstemperatur) ~ 102 bis 104 Glas (Raumtemperatur) ~ 1018 bis 1020 Blut (37 °C) 4 bis 25 Traubensaft 2 bis 5 Olivenöl ~ 102 Honig ~ 104 Sirup ~ 105 Kaffeesahne ~ 10 Steinsalz ~ 1013 bis 1018
1) Sofern nicht anders vermerkt, beziehen sich die Werte auf die Viskosität bei 20 °C.
2) Bei Polymeren gibt es einen sehr breiten Bereich an Viskositäten, der im Wesentlichen von der Kettenlänge und deren Verzweigungsstruktur (und natürlich von der Temperatur) abhängt. Hergestellt werden z.B. Siliconöle (PDMS) mit definierten Viskositäten zwischen 0,6 mPa·s bei 25 °C und 1.000.000 mPa·s bei 27 °C. Polymerschmelzen können aber auch noch sehr viel höhere Viskositäten aufweisen. Bei einem UHMW-HDPE (für Hüftgelenksimplantate) wurden Viskositäten jenseits der 1013 mPa·s bei 150 °C gemessen.Man muss aber bedenken, dass bei normalen ungefüllten Polymerschmelzen spätestens ab einer Viskosität von 10.000 mPa·s Strukturviskosität auftritt, deren Intensität (also wie stark die Viskosität bei hoher Scherrate sinkt) sich bei steigender Viskosität erhöht.
Bei der Landhebung, einem Nacheiszeitlichen Effekt der Erdoberfläche, sind es rund 1021 Pa·s.
Viskosität von Gasen
 Viskosität verschiedener Gase bei Normaldruck
Viskosität verschiedener Gase bei Normaldruck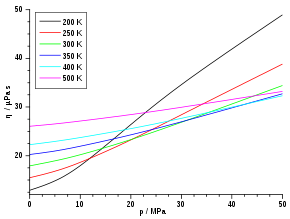 Viskosität von Stickstoff in Abhängigkeit vom Druck für verschiedene Temperaturen
Viskosität von Stickstoff in Abhängigkeit vom Druck für verschiedene Temperaturen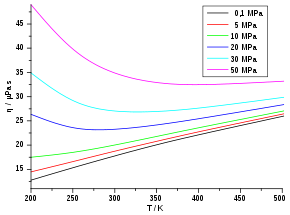 Viskosität von Stickstoff in Abhängigkeit von der Temperatur für verschiedene Drücke
Viskosität von Stickstoff in Abhängigkeit von der Temperatur für verschiedene DrückeFür Gase lässt sich die Viskosität anhand einer mikroskopischen Betrachtung des Impulsflusses abschätzen:
mit der freien Weglänge λ für die Gasteilchen, der Masse der Gasteilchen m, der mittleren Teilchengeschwindigkeit v und der Teilchenzahldichte n.
Die Viskosität von Gasen ist bei niedrigen Drücken (ca. 0,1 bis 10 bar) unabhängig vom Druck. Dies gilt solange, wie die freie Weglänge klein gegenüber den Gefäßabmessungen und groß gegenüber den Molekülabmessungen ist. Mit anderen Worten: Für ein sehr dünnes oder ein sehr dichtes Gas wird die Viskosität doch wieder vom Druck beziehungsweise der Dichte des Gases abhängig.
Grundsätzlich abhängig ist die Viskosität aber von der Temperatur. Mit zunehmender Temperatur steigt die Viskosität, da die mittlere Teilchengeschwindigkeit v proportional zu T0,5 wächst (vgl. Abschnitt „Kinetische Gastheorie“). Dieses Verhalten ist bei den meisten Flüssigkeiten genau entgegengesetzt. Die folgende Tabelle listet zu einigen Gasen die Viskositäten und freien Weglängen auf.
-
Gas η (273 K) in µPa·s λ (1 atm) in nm Luft 17,1 59,8 Sauerstoff (O2) 19,2 63,3 Kohlendioxid (CO2) 13,8 39,0 Stickstoff (N2) 16,6 58,8 Argon 21,0 62,6 Neon 29,7 124,0 Helium 18,6 174,0 Wasserstoff (H2) 8,4 111,0
Kinetische Gastheorie
Nach Hirschfelder kann die Viskosität reiner Gase mit Hilfe der kinetischen Gastheorie in einem großen Temperaturbereich (etwa von 200 bis 3000 Kelvin) berechnet werden.
Hierbei ist m die Molekülmasse, kB die Boltzmann-Konstante, T die Temperatur, σ der Lennard-Jones-Stoßdurchmesser und
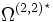 das reduzierte Stoßintegral, das von der reduzierten Temperatur
das reduzierte Stoßintegral, das von der reduzierten Temperatur 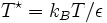 abhängt. ε ist die Energie des Lennard-Jones-Potentials. Werte für die Lennard-Jones-Parameter und das reduzierte Stoßintegral sind in Lienhards Lehrbuch zur Wärmeübertragung in Kapitel 11 aufgeführt. Das reduzierte Stoßintegral ist so definiert, dass für ein ideales Gas, bei dem Teilchenwechselwirkungen wie Stöße harter Kugeln betrachtet werden, gilt
abhängt. ε ist die Energie des Lennard-Jones-Potentials. Werte für die Lennard-Jones-Parameter und das reduzierte Stoßintegral sind in Lienhards Lehrbuch zur Wärmeübertragung in Kapitel 11 aufgeführt. Das reduzierte Stoßintegral ist so definiert, dass für ein ideales Gas, bei dem Teilchenwechselwirkungen wie Stöße harter Kugeln betrachtet werden, gilt 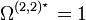 .
.Fluidität
Der Kehrwert der Viskosität ist die Fluidität η − 1 mit der Einheit
![[\eta^{-1}]=\frac{\rm m \cdot s}{\rm kg}](/pictures/dewiki/55/75f4062d0c0c0373c8907edca3dd40f0.png) .
.Siehe auch
- Gesetz von Stokes
- Navier-Stokes-Gleichungen
- Gesetz von Hagen-Poiseuille
- Engler-Grad
- Visco-Kupplung
- Rheologie
- Vulkameter
- Viskosimeter
- Andrade-Gleichung
- Vogel-Fulcher-Tammann-Gleichung
- Pechtropfenexperiment
Literatur
- Joseph O. Hirschfelder, Charles F. Curtiss, und Robert Byron Bird: Molecular Theory of Gases and Liquids. Wiley, 1964, ISBN 0-471-40065-3
- John H. Lienhard IV und John H. Lienhard V, A Heat Transfer Textbook. Phlogiston Cambridge, 3. Auflage, 2005
- Atkins, Peter W.: Physikalische Chemie / A. Höpfner (Übers.). 3., korr. Aufl. Weinheim, Wiley-VCH, 2002, ISBN 3-527-30236-0
- Dealy JM.: Structure and Rheology of Molten Polymers. München, Hanser Fachbuchverlag, 2006
- Gabriel C.: Einfluss der molekularen Struktur auf das viskoelastische Verhalten von Polyethylenschmelzen. Lehrstuhl für Polymerwerkstoffe, Erlangen, Friedrich-Alexander-Universität Erlangen-Nürnberg, 2001
- Piel C, Stadler FJ, Kaschta J, Rulhoff S, Münstedt H, Kaminsky W: Structure-property relationships of linear and long-chain branched metallocene high-density polyethylenes and SEC-MALLS. Macromolecular Chemistry and Physics 207 (1), 26-38, 2006
- Gehm, Lothar: RHEOLOGIE – Praxisorientierte Grundlagen und Glossar. VINCENZ 1998, ISBN 3-87870-449-6
- Schwarzl, FR.: Polymermechanik. Heidelberg, Berlin, New York, Springer, 1993
Weblinks

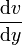
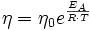
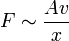
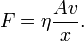
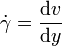

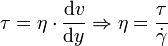
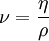





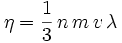
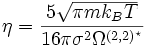
Wikimedia Foundation.