- Gaschromatografie
-
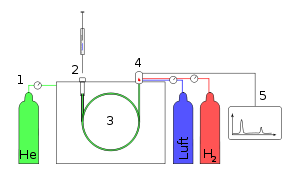 Prinzipieller Aufbau eines Gaschromatographen: Trägergas (1), Injektor (2) Säule im GC-Ofen (3), Detektor, hier FID (4), Signalaufzeichnung (5).
Prinzipieller Aufbau eines Gaschromatographen: Trägergas (1), Injektor (2) Säule im GC-Ofen (3), Detektor, hier FID (4), Signalaufzeichnung (5).Die Gas-Flüssigkeits-Chromatographie (GLC) oder einfach Gaschromatographie (GC) ist eine Verteilungschromatographie, die als Analysenmethode zum Auftrennen von Gemischen in einzelne chemische Verbindungen weite Verwendung findet. Die GC ist nur anwendbar für Komponenten, die gasförmig sind oder verdampfen (Siedebereich bis 400°C). Bei dieser Art der Chromatographie wird als mobile Phase ein Inertgas wie Stickstoff oder Helium verwendet, in besonderen Fällen auch Wasserstoff. Das Trägergas wird durch eine gebogene, gewickelte, kapillarartige Röhre, die sogenannte Säule, die häufig eine Länge zwischen 10-200 Meter besitzt, gedrückt. Die Säule besteht entweder aus Metall (bei älteren Säulen) oder aus einem zur Erhöhung der Bruchsicherheit beschichteten Quarzglas (heutzutage die Regel) und befindet sich in einem temperierbaren Ofen. Sie ist innen mit einer definierten stationären Phase ausgekleidet, häufig mit zähflüssigen Polyorganosiloxanen. Nach Eingabe einer Probesubstanz, die nun vom Trägergas mitgerissen wird, verweilen die Komponenten je nach Polarität der einzelnen Gasmoleküle unterschiedlich lange an der stationären Phase der Säule. Mit einem Detektor kann man den Austrittszeitpunkt am Säulenende messen. Mit einem Schreiber, der am Detektor angebracht ist, kann der Zeitpunkt des Austritts am Rohrende und die Menge der Substanz graphisch dargestellt und mit Standardsubstanzen verglichen werden. Damit ist eine sehr schnelle und leichte qualitative und quantitative Bestimmung selbst von sehr komplexen Stoffgemischen möglich. Im Unterschied zur HPLC sind nur ausreichend flüchtige Substanzen nachweisbar.
Inhaltsverzeichnis
Wichtige Geräteteile
Ein Gaschromatograph besteht aus drei wesentlichen Bauteilen: Injektor, Trennsäule im GC-Ofen und Detektor. Im Injektor wird die Probe, gelöst in einem niedrig siedenden Lösemittel, durch ein Septum eingespritzt. Dieser Injektor wird in der Regel beheizt (bis zu 450 °C), um eine rasche und vollständige Verdampfung der Probe zu erreichen. Möglich ist auch die septumfreie Aufgabe und langsame Verdampfung mittels eines Kaltaufgabesystems (KAS/PTV). Die Substanzen werden durch das Trägergas (Säulenvordruck normalerweise bis zu 6 bar) in die Trennsäule (Kapillare) transportiert, welche in den so genannten GC-Ofen eingebaut ist. Dieser dient dazu, die Trennsäule präzise zu temperieren, um so durch konstante Temperatur (isotherm) oder durch Temperaturgradienten, d.h. durch kontrollierte Temperaturerhöhung, eine ebenso schnelle wie weitgehende Trennung des Stoffgemisches zu erreichen. Am Ende der Säule folgt der Detektor, der ein elektronisches Signal erzeugt, wenn eine Substanz das Trennsystem verlässt. Das elektronische Signal kann als Peak (engl. Gipfel) auf dem Schreiber registriert werden. Die Signale werden dann an einem Integrator - oder heute meist an einem Computersystem mit entsprechender Auswertesoftware - verarbeitet. Die Dauer für die Trennung eines Stoffgemisches mit der Darstellung der verschiedenen Peaks zu einem Chromatogramm beträgt ca. 30-60 Minuten.
Messprinzip
Die chromatographische Auftrennung eines Stoffgemisches in einem Gaschromatographen erfolgt bei einer unpolaren Trägersäule im einfachsten Falle ausschließlich aufgrund der unterschiedlichen Siedepunkte der Einzelsubstanzen in dem Gemisch, wobei keine spezielle Wechselwirkung mit der stationären Phase erfolgt, sondern "nur" eine zehntausendfach wiederholte Verteilung. Bei polaren Trennsäulen werden aber Alkohole, Ester, Ketone mit gleichen Siedepunkten wie ähnliche Paraffine stärker festgehalten. Die spezielle Wechselwirkung - genauer das Gleichgewicht zwischen der Gasphase und der stationären Phase - ist als Raoultsches Gesetz bekannt. Je höher der Partialdampfdruck, einer Substanz nach dem Raoultschen Gesetz, d.h. je länger sich die Substanz in der Gasphase befindet, desto kürzer wird die Retentionszeit.
Die Stärke der Wechselwirkungen, zwischen den Probenkomponenten und der stationären Phase, wird sowohl von deren Struktur als auch von deren funktionellen Gruppen bestimmt. Dabei treten bei unpolaren Substanzen ausschließlich Dispersionswechselwirkungen (Van-der-Waals-Bindungen) auf, während polare Trennphasen auch polare Wechselwirkungen eingehen können, z.B. Wasserstoffbrückenbindungen oder Donator-Akzeptor-Bindungen. Letztere trennen nach dem Prinzip: Gegensätze ziehen sich an. Das bedeutet, dass Trennphasen, die z.B. Wasserstoff zur Wasserstoffbrückenbindung aufzunehmen in der Lage sind, Substanzen trennen, die Wasserstoff zur Brückenbindung bereitstellen können (z.B. Alkohole). Auch können zum Beispiel Enantiomere, welche sich in ihren Siedepunkten nicht unterscheiden und somit gleiche Retentionszeiten aufweisen würden, durch ihre verschieden starken Wechselwirkungen mit speziellen Derivaten von Cyclodextrinen aufgetrennt werden.
Eine Grundbedingung für die Gaschromatographie ist, dass sich der Stoff, den man untersuchen möchte, unzersetzt verdampfen lässt - sofern er nicht schon gasförmig vorliegt. Mittels Derivatisierung lassen sich der GC ansonsten schwer zugängliche Analyten wie Alkohole, Amine, Fettsäuren oder Zucker soweit thermisch stabilisieren, dass sie ohne Schwierigkeiten auf handelsüblichen Phasen aufgetrennt werden können. Mögliche Derivate sind bei Carbonsäuren die Methylester (Umwandlung mit BF3 und MeOH), bei Alkoholen die Silylether.
Injektoren
Der Injektor dient der Aufgabe des zu untersuchenden Stoffgemisches auf die Trennsäule. Gängige Injektoren / Methoden sind:
- Split/Splitless-Injektoren (SSL)
- On Column-Injektoren (OCI) mit Direktaufgabe auf die Säule
- Kaltaufgabesysteme (je nach Hersteller KAS oder PTV)
- Direktaufgabesysteme mittels Ventilschaltungen
Für gepackte Säulen beträgt die optimale Menge der Probe je Komponente zwischen 0,1 – 1 μl, für Kapillarsäulen sollte die optimale Probemenge um den Faktor 100 bis 1000 kleiner sein. Zur Injektion einer Probe, die man auch durch ein Lösungsmittel noch verdünnen kann, gibt es spezielle 1-10 μl Spritzen. Wichtig für die Injektion ist, dass sich keine Luft (-blasen) in der Spritze befindet, diese würde nämlich zu einer Oxidation der Substanzen im Ofenraum beitragen.
Gerade mit Split/Splittless-Injektoren und Kaltaufgabesystemen werden häufig sogenannte Autosampler eingesetzt, die die sequentielle Abarbeitung einer Vielzahl an Proben erlauben. Daneben werden u.a. auch Headspace-Probengeber, Purge&Trap-Systeme und Pyrolysatoren zur Probenaufgabe verwendet. Eine recht neue Entwicklung ist der Einsatz von Festphasenmikroextraktion (SPME).
Verwendete Trennsäulen
Wichtige Kenngrößen der Trennsäulen sind:
- der Säulendurchmesser und
- die Säulenlänge.
Früher wurden meist gepackte Säulen eingesetzt. Bei gepackten Säulen befindet sich im Inneren eines dünnen (<1 cm) Metall- oder Glasrohres von wenigen Metern Länge, der sogenannten Säule, ein festes, inertes Trägermaterial. Wird das Gas mit der zu analysierenden Substanz direkt über das Trägermaterial geleitet, so spricht man von einer GSC ("Gas-Solid-Chromatography"). Ist die Trägersubstanz zudem mit einer dünnen Schicht einer hochmolekularen, zähflüssigen und wenig flüchtigen Flüssigkeit überzogen, so handelt es sich um eine GLC ("Gas-Liquid-Chromatography"). Diese Flüssigkeit übernimmt hier die Funktion der eigentlichen stationären Phase. Das für eine Säulenchromatographie bevorzugt verwendete Inertgas ist Stickstoff.
Heutzutage wird überwiegend mit der von Golay 1958 entwickelten Kapillarchromatographie gearbeitet. Der Vorteil besteht in der ca. 100-1000 fach besseren Auftrennung, (einer Trennstufenzahl von ca. 300.000) von Stoffen verglichen mit gepackten Säulen, so dass sich auch die Analysezeit verkürzen läßt. Dabei haben die mit Polyimid beschichteten Quarzglas-Trennsäulen normalerweise einen Innendurchmesser von 0,1 bis 0,5 mm und eine Länge von 10 bis 60 m. Zur Auftrennung von Fettsäureestern werden sogar kombinierte Säulen bis 100 m verwendet. Die stationäre Phase kleidet die Kapillare dabei nur als dünner Film aus. Der Vorteil besteht in der drastisch besseren Auftrennung ähnlicher Stoffe, verglichen mit gepackten Säulen. Der Trend in der GC geht momentan zu immer dünneren und kürzeren Säulen, weil dadurch der Zeitaufwand für Analysen deutlich gesenkt werden kann.
Bei der Verwendung von Säulen unterschiedlicher Hersteller ist zu beachten, dass identische stationäre Trennphasen mit den unterschiedlichsten Bezeichnungen versehen werden. Der folgenden Liste gängiger stationäre Trennphasen ist zu entnehmen, welche Trennsäulen unterschiedlicher Hersteller hinsichtlich Zusammensetzung der Belegung der Trennsäulen vergleichbar sind.
Zusammensetzung der Belegung der
TrennsäuleHerstellerbezeichnung Temperaturbereich Polydimethylsiloxan DB-1, SB-1, BP-1, CP-Sil 5 CB, −50 °C bis +350 °C 100 % Methyl PB-1, SPB-1, RTX-1, PE-1, Ultra 1, AT-1, SE-30 Polyphenylmethylsiloxan DB-5, SB-5, BP-5, CP-Sil 8 CB, PVMS-5 −20 °C bis +350 °C 5 % Phenyl, 95% Dimethyl PB-2, SPB-5, Rtx-5, PE-2, Ultra 2, AT-5, SE-54, Optima-5 RSL-200 Polycyanopropylphenylsiloxan DB-225, SB-225, BP-15, CP-Sil 43 CB, ±0 °C bis +250 °C 25% Cyanopropyl, 25% Phenylmethyl RTX-225, PE-225, HP-225, AT-225, RSL-500 Polyphenylmethylsiloxan DB-624, SB-624, CP-Sil 13 CB, ±0 °C bis +250 °C 14%Phenyl, 86% Dimethyl VOCOL, Rtx-Volatiles, PE-502, AT-62 - Polycyanopropylphenylmethylsiloxan DB-1301, SB-1301, Rtx-1301, ±0 °C bis +230 °C 6% Cyanopropylphenyl, 94% Dimethyl AT-1301 - Polyphenylmethylcyanosiloxan DB-1701, SB-1701, BP-10, −50 °C bis +225 °C 6% Phenyl, 6% Cyano, 88% Methyl CP-Sil 19 CB, PB-1701, SPB-7, Rtx-1701, PE-1701, PAS-1701, AT-1701, RSL-300 Polyethylenglykol DB-WAX, SB-Wax, BP-20, CP-Wax 52 CB, ±0 °C bis +220 °C Supelcowax-10, Stabilwax, PE-CW, HP-20M, AT-Wax Detektoren
 Gaschromatograph.
Gaschromatograph.Folgende Detektoren werden eingesetzt:
- Flammenionisationsdetektor (FID), allgemein für die Quantifizierung organischer Verbindungen
- Wärmeleitfähigkeitsdetektor (WLD o. TCD), für Permanentgase
- Photoionisationsdetektor (PID)
- Flammenphotometrischer Detektor (FPD), elementspezifisch
- Stickstoff-Phosphor-Detektor (NPD), Stickstoff-Phosphor-spezifisch
- Elektroneneinfangdetektor (ECD), für halogenierte organische Verbindungen
- Atomemissionsdetektor (AED), elementspezifisch
- Thermionischer Detektor (TID)
- Massenspektrometer, massenselektiver Detektor
- Ionen-Mobilitäts-Spektrometer (IMS) für flüchtige organische Verbindungen
Teilweise werden für spezielle Fragestellungen auch zwei Detektoren hintereinander geschaltet (Tandem-Prinzip). Grundvoraussetzung dafür ist aber, dass der erste Detektor seine Messung nicht zerstörend durchführt (also kein FID/NPD/PID, sondern ein ECD/WLD).
Am Detektor ist noch eine elektronische Auswerteeinheit (Computer) angebracht, die die zeitliche Darstellung (Retentionszeit) durch ein Chromatogramm ermöglicht.
Anwendung in der Analytik
Die Gaschromatographie ist eine sehr empfindliche Methode zur Analyse von Stoffgemischen. Es lassen sich selbst minimale Substanzmengen (10-9 Gramm) nachweisen. Man kann mit ihr komplexe Stoffgemische in die einzelnen Komponenten auftrennen. In vielen Fällen reicht allein die Zeit, die eine Substanz vom Zeitpunkt der Einspritzung bis zum Passieren des Detektors benötigt, die Retentionszeit, um eine Substanz zu identifizieren. Durch Kombination mit einem Massenspektrometer, die sogenannte GC/MS-Kopplung, können sehr geringe Substanzmengen nachgewiesen werden, und gleichzeitig Strukturaufklärung betrieben werden.
Anwendung findet die Gaschromatographie in der Analytik von Agrarprodukten auf Herbizide, Fleischprodukte auf Hormone, der Untersuchung von Arzneimittel, von Aromen und Etherischen Ölen, von Kohlenhydraten, von Erdölkomponenten und in der forensischen Chemie, bei Dopingtests, bei Luft- und Meerwasseruntersuchungen in der Umweltanalytik .
Verwendung in der quantitativen Analyse
Die vom Detektor angezeigten Flächenprozent stimmen in den seltensten Fällen mit den tatsächlichen Gewichtsprozent überein. Abgesehen davon sind vor allem ältere Split-Injektoren nicht reproduzierbar, weshalb eine Kalibrierung notwendig ist. Diese wird mittels eines internen Standards vorgenommen. Dabei wird eine zusätzliche Substanz, deren Retetionszeit in der Nähe der zu bestimmenden Substanzen liegt, aber diese nicht überlagern, benötigt. Sie wird einmal dem zu messenden Gemisch beigesetzt, und einmal der Referenzsubstanz. Nach der Messung können die Peaks mit dem Internen Standard geglättet werden. Nach erfolgter Kalibrierung erlaubt die integrierte Peakfläche der Substanz einen Rückschluss auf ihren Massenanteil in der Probe.
Literatur
- Gerhard Schomburg: Gaschromatographie, Grundlagen, Praxis, Kapillartechnik. Verlag Chemie, Weinheim 1987.
- Peter J. Baugh: Gaschromatographie. Eine anwenderorientierte Darstellung. Springer, Berlin 1997. ISBN 3540670092
- Walter David: GC-Tipps. Hoppenstedt Bonnier Zeitschriften (1999). ISBN 3935772033
- Bruno Kolb: Gaschromatographie in Bildern. ISBN 3-527-29880-0
- Ullmanns Encyclopädie der technischen Chemie, 4. Auflage, Band 5, S. 118 ff.
- Leibniz, Eberhardt; Struppe, Hans-Georg: Handbuch der Gaschromatographie. Akademische Verlagsgesellschaft Geest&Portig KG, Leipzig 1984
Weblinks


Wikimedia Foundation.