- Glioblastoma multiforme
-
Klassifikation nach ICD-10 C71 Bösartige Neubildung des Gehirns C71.0 Zerebrum, ausgenommen Hirnlappen und Ventrikel C71.1 Frontallappen C71.2 Temporallappen C71.3 Parietallappen C71.4 Okzipitallappen C71.5 Hirnventrikel C71.6 Zerebellum C71.7 Hirnstamm C71.8 Gehirn, mehrere Teilbereiche überlappend C71.9 Gehirn, nicht näher bezeichnet ICD-10 online (WHO-Version 2006) Das Glioblastom (medizinisch korrekt: Glioblastoma multiforme) ist der häufigste bösartige Hirntumor bei Erwachsenen. Das Glioblastom weist feingewebliche Ähnlichkeiten mit den Gliazellen des Gehirns auf und wird aufgrund der sehr schlechten Prognose nach der WHO-Klassifikation der Tumoren des zentralen Nervensystems als Grad IV eingestuft. Die Behandlung besteht in operativer Reduktion der Tumormasse, Bestrahlung und Chemotherapie. Eine endgültige Heilung kann derzeit nicht erreicht werden. Die mittlere Überlebenszeit liegt in der Größenordnung von Monaten, manche Erkrankte überleben auch länger, nur einzelne jedoch mehrere Jahre.
Inhaltsverzeichnis
Historisches
Der Begriff Glioblastoma multiforme wurde 1926 von Percival Bailey und Harvey Cushing geprägt und basierte auf der Vorstellung, dass sich der Tumor aus primitiven Vorstufen von Gliazellen (Glioblasten) entwickelt sowie der Beobachtung, dass das Erscheinungsbild mit Nekrosen, Einblutungen und Zysten sehr variabel (multiform) sein kann.[1] Der von dem Pathologen Frank Burr Mallory bereits 1914 verwendete Begriff Spongioblastoma multiforme[2] konnte sich nicht durchsetzen.
Verbreitung
Etwa ein Fünftel aller hirneigenen Tumore sind Glioblastome, unter den Gliomen machen sie etwas mehr als die Hälfte aus. Der Tumor tritt am häufigsten bei älteren Erwachsenen auf; das mediane Alter bei Diagnosestellung beträgt 64 Jahre. Jenseits des 45. Lebensjahres ist es nach dem Meningeom der häufigste Hirntumor. Männer sind deutlich öfter betroffen als Frauen (Verhältnis 1,7:1). Daten des amerikanischen Hirntumorregisters zeigen, dass Glioblastome bei Weißen mindestens doppelt so häufig sind wie in der schwarzen Bevölkerung. Im Vergleich zu Erwachsenen sind Glioblastome bei Kindern sehr selten. Die Inzidenz wurde in Europa und Nordamerika mit 2,9 bis 3,5 Neuerkrankungen pro Jahr auf 100.000 Einwohner ermittelt.[3][4][5] Als einer der wenigen bekannten Risikofaktoren ist eine hohe Dosis ionisierender Strahlung anzusprechen.
Bei der Mehrzahl der Glioblastome handelt es sich um sporadisch auftretende Fälle ohne Hinweis auf eine Erblichkeit. Beim Li-Fraumeni-Syndrom oder dem Turcot-Syndrom, seltenen erblichen Erkrankungen, können Glioblastome jedoch familiär gehäuft auftreten.
Krankheitsentstehung
Glioblastome können völlig neu (de novo) oder durch fortschreitende Entdifferenzierung aus weniger bösartigen Astrozytomen entstehen. Daher kommt es nicht selten vor, dass therapierte Astrozytome sich im Rezidiv als Glioblastom manifestieren. Diese sogenannten sekundären Glioblastome treten eher bei jüngeren Patienten auf und haben ein anderes Spektrum genetischer Veränderungen als neuentstandene (siehe Molekularpathologie). In einer in der Schweiz durchgeführten epidemiologischen Studie waren primäre Glioblastome im Kanton Zürich etwa 20mal häufiger als sekundäre.[5]
Lokalisation
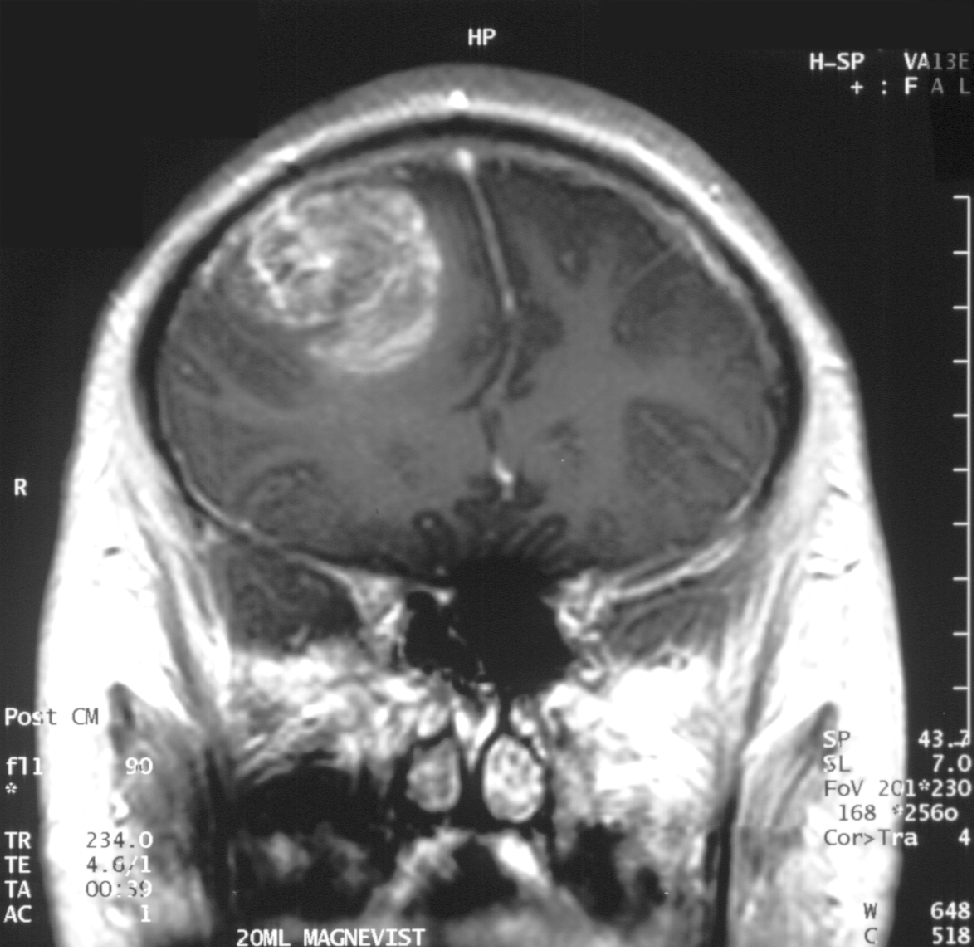
 Coronales MRT mit Kontrastmittel eines Glioblastoms bei einem 15 Jahre alten Jungen. Deutlich ist der raumfordernde Effekt (Herniation) an der Verlagerung der Mittellinie (Falx cerebri) erkennbar.
Coronales MRT mit Kontrastmittel eines Glioblastoms bei einem 15 Jahre alten Jungen. Deutlich ist der raumfordernde Effekt (Herniation) an der Verlagerung der Mittellinie (Falx cerebri) erkennbar.Das Glioblastom geht von der weißen Substanz aus. Seine mit Abstand häufigste Lokalisation ist das Großhirn, wo es in allen Hirnlappen entstehen kann, aber den Frontal- und den Temporallappen bevorzugt. Im Bereich von Kleinhirn, Hirnstamm und Rückenmark sind Glioblastome selten. Oft wachsen hemisphärielle Glioblastome über den Balken auf die andere Seite hinüber. Solche Tumoren werden als sogenannte „Schmetterlingsgliome“ bezeichnet. Das Wachstum von Glioblastomen ist diffus infiltrierend.
Klinische Erscheinungen
Wegen des raschen Wachstums entwickeln sich die Beschwerden meistens rasch innerhalb weniger Wochen bis Monate. Erste Symptome können anhaltende und ungewohnte Kopfschmerzen, aber auch neu auftretende epileptische Anfälle sein. Fokale neurologische Ausfälle wie Lähmungen, Aphasien und Sehstörungen können lokalisationsabhängig hinzukommen. Schließlich sind es oft auffällige Persönlichkeitsveränderungen, Apathie oder psychomotorische Verlangsamung, die den Patienten zum Arzt führen. Hirndruckzeichen wie Stauungspapille, Erbrechen, Somnolenz und Koma treten spät auf und sind prognostisch ungünstig.
Untersuchungsmethoden
Die Diagnose wird zunächst durch bildgebende Verfahren (Computertomographie (CT), Magnetresonanztomographie (MRT)) gestützt. In der CT-Bildgebung mit Kontrastmittel erscheint das Glioblastom unregelmäßig geformt mit randständig starker Kontrastmittelaufnahme (ringförmiges Enhancement). Bei kleineren Tumoren ist dieses ringförmig konfiguriert, bei größeren bildet es eine girlandenartige Formation aus. In der Umgebung des Tumors bildet sich typischerweise ein erhebliches Ödem aus. Der MRT-Befund ist recht typisch: die soliden Anteile des Glioblastoms reichern Kontrastmittel stark an, dagegen heben sich die Aussparungen durch zystische Anteile und die Blutungen ab. Letztendlich wird die Diagnose am Tumorgewebe, das bei einer stereotaktischen Hirnbiopsie oder Tumorresektion gewonnen wurde, neuropathologisch bestätigt. Im Einzelfall werden Supplementäruntersuchungen wie Elektroenzephalografie und Lumbalpunktion durchgeführt, die der Einschätzung der Anfallsneigung bzw. der differentialdiagnostischen Abgrenzung gegen Hirnabszesse oder Lymphome dienen.
Pathologie
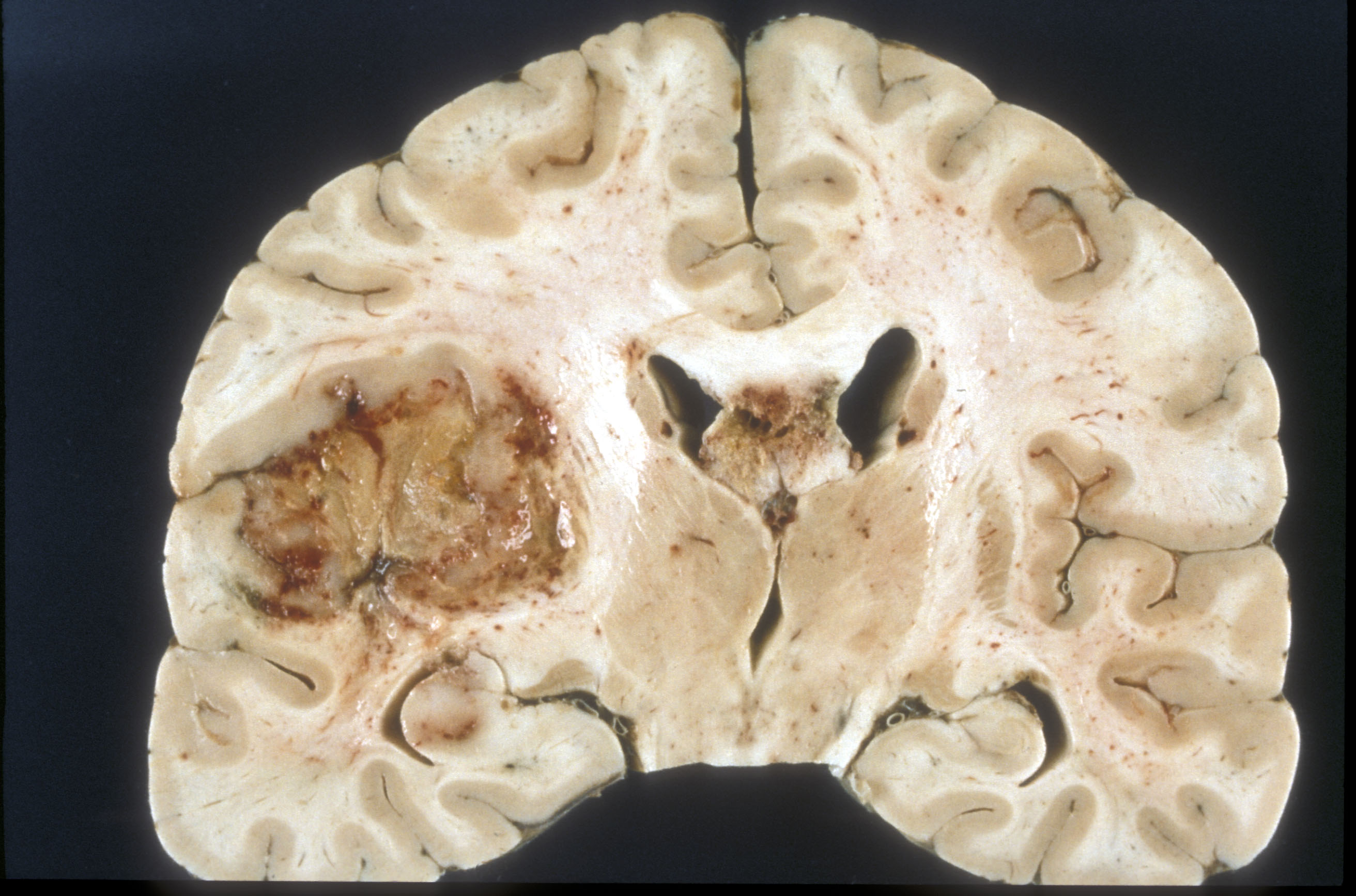
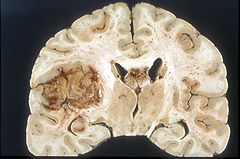 Glioblastom (Makroskopisches Präparat). Koronare Schnittfläche eines formalinfixierten Gehirns). Der Tumor stellt sich als grau-roter, teils nekrotischer Bereich des linken Schläfen- und Frontallappens dar. Der Tumor hat sich außerdem in den Balken ausgebreitet (Mitte des Bildes, dunkelgrauer Bereich.
Glioblastom (Makroskopisches Präparat). Koronare Schnittfläche eines formalinfixierten Gehirns). Der Tumor stellt sich als grau-roter, teils nekrotischer Bereich des linken Schläfen- und Frontallappens dar. Der Tumor hat sich außerdem in den Balken ausgebreitet (Mitte des Bildes, dunkelgrauer Bereich.Das Glioblastom ist durch seine inhomogene und vielfältige (daher: multiforme) Erscheinung gekennzeichnet: die Tumorschnittfläche weist häufig rötliche Einblutungen und gelbliche Gewebsuntergänge (Nekrosen) auf.
Histologie
.jpg) Glioblastom (Histologisches Präparat mit typischen strichförmigen Nekrosen und palisadenartiger Anordnung pleomorpher Tumorzellen um die Nekrosen.) (Hämatoxylin-Eosin-Färbung)
Glioblastom (Histologisches Präparat mit typischen strichförmigen Nekrosen und palisadenartiger Anordnung pleomorpher Tumorzellen um die Nekrosen.) (Hämatoxylin-Eosin-Färbung)Feingeweblich (histologisch) handelt es sich um zelldichte, astrozytär differenzierte Tumoren, die diffus das umgebende reaktiv veränderte Hirngewebe infiltrieren. Die Tumorzellen sind mit multipolaren feinen Fortsätzen fibrillär-astrozytär differenziert oder weisen mit einem aufgeblähten Zytoplasma eine gemästet-zellige Differenzierung auf. Auch Riesenzellen mit bizarren Kernen oder kleinzellige Areale mit wenig ausgedehnten Zellkörpern kommen vor. Die Zellkerne sind meist chromatinreich und vielgestaltig (polymorph). Die mitotische Aktivität ist erhöht, der Proliferationsmarker KI-67 beträgt zum Teil mehr als 20 %.
Entscheidend für die Diagnose des Glioblastoms (und die Abgrenzung gegenüber anaplastischen Astrozytomen) ist nach der Tumorklassifikation der Weltgesundheitsorganisation jedoch der Nachweis von Tumornekrosen (flächenhaft oder typischerweise strichförmig mit perifokaler Zelldichtesteigerung) oder hochgradig pathologischer Blutgefäße.
Varianten
Bei Gliosarkomen handelt es sich um Glioblastome, die neben den oben beschriebenen astrozytären Tumoranteilen auch bindegewebsreiche sarkomatöse Abschnitte mit spindelzelligen Tumorzellen aufweisen. Als Riesenzellglioblastome werden Glioblastome mit einer ausgeprägten riesenzelligen Komponente bezeichnet. Ebenfalls abzugrenzen sind Glioblastome mit oligodendroglialer Komponente, die möglicherweise eine etwas günstigere Prognose haben.[6]
Immunhistochemie
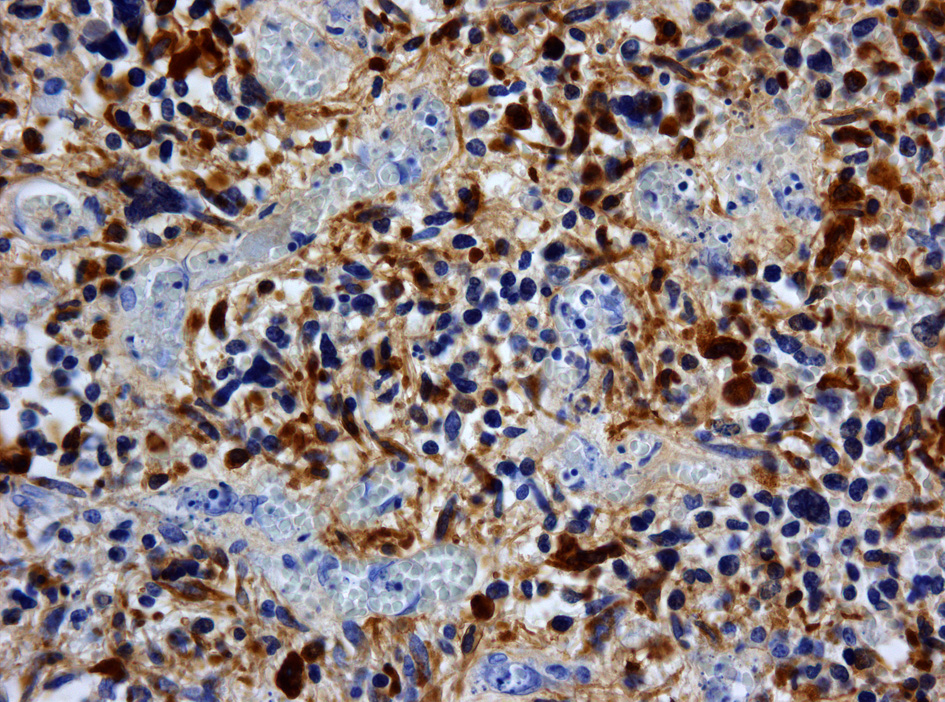
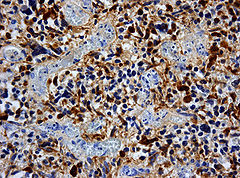 Immunhistochemische Färbung der Tumorzellen für GFAP
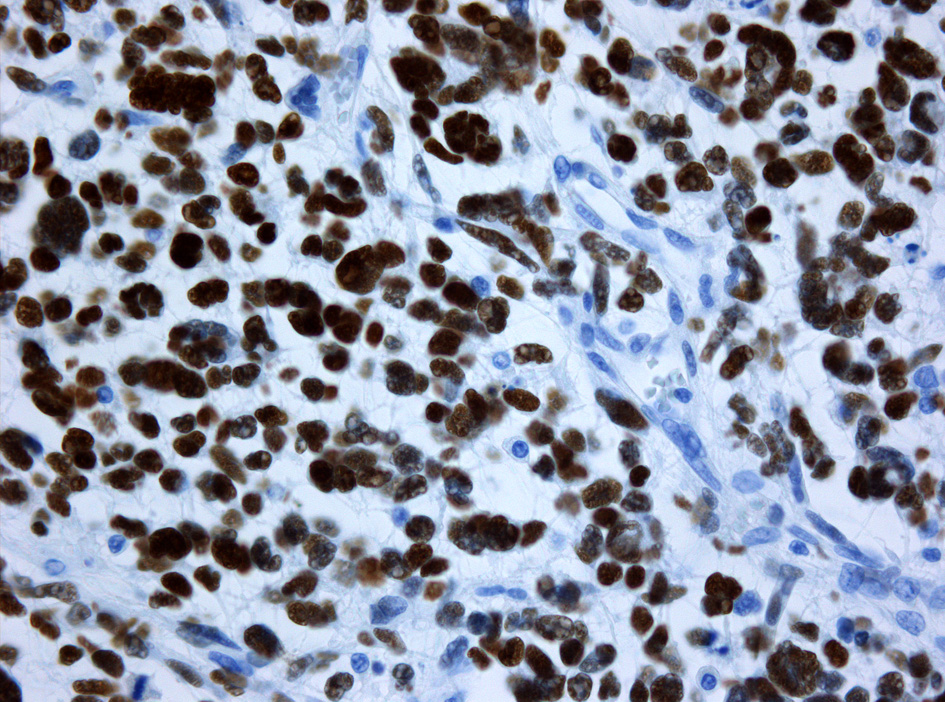
Immunhistochemische Färbung der Tumorzellen für GFAP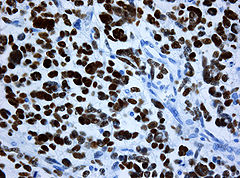 Immunhistochemische Färbung der Tumorzellen für p53. Ansammlung von (defektem) p53-Protein in den Tumorzellkernen bei einem sekundären Glioblastom mit Mutation des TP53 Gens. Die Kerne mitgetroffener Blutgefäßwandzellen sind ungefärbt
Immunhistochemische Färbung der Tumorzellen für p53. Ansammlung von (defektem) p53-Protein in den Tumorzellkernen bei einem sekundären Glioblastom mit Mutation des TP53 Gens. Die Kerne mitgetroffener Blutgefäßwandzellen sind ungefärbtImmunhistochemisch ist in den Tumorzellen – wie in denen anderer glialen Hirntumoren – das gliale saure fibrilläre Protein (glial fibrillary acidic protein, GFAP) nachweisbar, was unter anderem die Abgrenzung gegenüber Hirnmetastasen erlaubt.[7]
Molekularpathologie
Die Genverluste (Deletionen), die das Glioblastom ausmachen, betreffen in den meisten Fällen das Tumorsuppressionsgen TP53 (Chromosom 17), das Retinoblastom-Suppressorgen RB-1 (Chromosom 13) und Deletionen des Chromosoms 22 sowie den Komplettverlust des Chromosoms 10. Diese genetischen Schäden liegen häufig kombiniert vor.
Bei neu entstandenen primären Glioblastomen, die überwiegend bei älteren Patienten auftreten, treten häufiger Verluste des PTEN-Gens oder eine Amplifikation des EGFR-Gens auf.[8]
Bei den überwiegend im mittleren Lebensalter auftreten sekundären Glioblastomen, welche durch eine schrittweise Fortentwicklung (Progression) aus weniger bösartigen (malignen) Astrozytomen entstandenen sind, liegen häufig Mutationen des TP53-Gens vor. Zudem zeichnet sich ab, das Punktmutationen des für eine Isocitrat-Dehydrogenase kodierenden IDH1-Gens in dieser Gruppe sehr häufig sind.[9]
Die seltenen kindlichen Glioblastome unterscheiden sich im Muster genetischer Veränderungen von den bei Erwachsenen auftretenden Tumoren.[10]
Behandlung
Eine kurzfristige klinische Besserung kann durch Behandlung des praktisch immer vorhandenen Hirnödems mit Dexamethason erreicht werden. Die neurochirurgische Operation mit Verminderung (Reduktion) der Hauptmasse des Tumors kann das Fortschreiten der Erkrankung verlangsamen, aber nicht dauerhaft verhindern, da praktisch immer einzelne Tumorzellen das gesunde Gehirngewebe schon infiltrativ durchwandert haben und deswegen eine vollständige Tumorentfernung nicht möglich ist. Zur Verlängerung der rezidivfreien und absoluten Überlebenszeit schließt sich deswegen an die Operation praktisch immer eine Bestrahlung und häufig auch eine Chemotherapie an, wobei insbesondere Patienten mit Nachweis epigenetischer Veränderungen (Hypermethylierung) des Promotors des DNA-Reparaturenzyms O6-Methylguanin-DNA-Methyltransferase (MGMT) von einer Chemotherapie mit Temozolomid profitieren.[11]
Weitere Chemotherapeutika, die unter anderem beim Rezidiv eingesetzt werden, sind Nitrosoharnstoffe, Vinkaalkaloide und Cytosinarabinosid, wobei verschiedene Schemata in Gebrauch sind.
Welche Patienten von einer lokalen Chemotherapie mit Implantation von an Polymere gebundenem Carmustin profitieren können, bleibt zu klären.[12]
Klinische Studien
Die Entwicklung neuer Behandlungsformen bei Glioblastomen ist Gegenstand intensiver Forschung. Im April 2009 waren 196 klinische Studien bei Clinicaltrials.gov, einem Register der United States National Library of Medicine als aktiv oder in Vorbereitung registriert.[13]
Tyrosinkinaserezeptoren wie die Rezeptoren für epidermalen Wachstumsfaktor (EGFR) und Platelet Derived Growth Factor (PDGF) stellen mögliche Zielmoleküle für neue therapeutische Ansätze dar.[14][15]
Eine Behandlung mit Bevacizumab, einem den Vascular endothelial growth factor (VEGF) neutralisierenden Antikörper, konnte in klinischen Studien in Kombination mit dem Topoisomerasehemmer Irinotecan die Tumorausdehnung reduzieren, wobei einzelne Patientengruppen möglicherweise besonders günstig auf diese Behandlung ansprechen.[16]
Auch gentherapeutische Verfahren werden im Rahmen klinischer Studien erprobt.[17]
Tierexperimentelle Studien
Ein beispielhafter experimenteller Ansatz ist die Behandlung mit Nanoteilchen.[18] Diese bestehen aus einem Eisenoxidkern sowie einer Hülle, die das Eindringen der Partikeln in die Krebszellen erleichtern soll. Die Partikel werden direkt in den Tumor injiziert. In mehreren Durchgängen wird der so mit den Eisenoxidpartikeln angereicherte Tumor mit Magnetwechselfeldern auf über 46 °C erwärmt. Im Tiermodell ergaben sich deutlich verbesserte Überlebenszeiten[19], Ergebnisse randomisierter kontrollierter Studien beim Menschen liegen noch nicht vor.
Prognose
Das Glioblastom ist äußerst schwierig zu behandeln. Eine endgültige Heilung ist bislang nicht möglich. Die optimale medizinische Behandlung mit Operation sowie nachfolgender Bestrahlung und Chemotherapie kann nach aktueller Studienlage die mittlere Überlebenszeit um einige Monate verlängern und die Symptome lindern, die 5-Jahres-Überlebensrate liegt jedoch nur um 3 %.[4] Wegen der diffusen Infiltration des Hirngewebes durch Tumorzellen kommt es nach der Behandlung häufig innerhalb von Monaten zu einem Rezidiv. Einzelne Patienten können dessen ungeachtet mehrere Jahre bei relativ guter Gesundheit mit einem Glioblastom leben. Die Identifizierung klinischer und molekularer Faktoren, die charakteristisch für solche Langzeitüberlebenden sind, ist Gegenstand intensiver Forschung.[20]
Literatur
Wolfgang Wick, Jörg-Christian Tonn, Michael Weller: Primäre intrakranielle und spinale Tumoren. In: Thomas Brandt, Johannes Dichgans und Hans Christoph Diener (Hrsg.): Therapie und Verlauf neurologischer Erkrankungen. 5. Auflage, Kohlhammer, Stuttgart 2007, ISBN 978-3-17-019074-0.
Einzelnachweise
- ↑ Bailey & Cushing: Tumors of the Glioma Group JB Lippincott, Philadelphia, 1926.
- ↑ Mallory: Principles of pathologic histology. Saunders Philadelphia, 1925.
- ↑ W.K. Cavenee et al.: Glioblastoma, in: WHO Classification of Tumours. Lyon, IARC Press, 2000.
- ↑ a b Amerikanisches Hirntumorregister
- ↑ a b H. Ohgaki, P. Kleihues: Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol 2005, 64(6):479-89; PMID 15977639.
- ↑ Homma et al.: Correlation among pathology, genotype, and patient outcomes in glioblastoma. J Neuropathol Exp Neurol. 2006 Sep, 65(9), 846–854; PMID 16957578.
- ↑ Velasco ME et al.: Immunohistochemical localization of glial fibrillary acidic protein in human glial neoplasms Cancer. 1980, 45, 484–494; PMID 6243508.
- ↑ Ohgaki H et al.: Genetic pathways to glioblastoma: a population-based study. Cancer Res 2004, 64, 6892–6899; PMID 15466178.
- ↑ Watanabe et al.:IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol. 2009;174(4):1149-53. PMID 19246647
- ↑ Rickert CH et al.: Pediatric high-grade astrocytomas show chromosomal imbalances distinct from adult cases. Am J Pathol 2001, 158, 1525–1532; PMID 11290570.
- ↑ Stupp et al.: Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005, 352, 987–996; PMID 15758009.
- ↑ Perry et al.: Gliadel wafers in the treatment of malignant glioma: a systematic review. Curr Oncol 2007, 14(5), 189–194; PMID 17938702 (Meta-Analyse); Volltext.
- ↑ [1] Abfrage bei clinicaltrials.gov am 22.04.2009
- ↑ Mellinghoff et al.: Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 2005, 353(19), 2012–2024; PMID 16282176.
- ↑ Reardon et al.: Phase II study of imatinib mesylate plus hydroxyurea in adults with recurrent glioblastoma multiforme. J Clin Oncol 2005, 23(36), 9359–9368; PMID 16361636.
- ↑ Sathornsumetee et al.: Tumor angiogenic and hypoxic profiles predict radiographic response and survival in malignant astrocytoma patients treated with bevacizumab and irinotecan. J Clin Oncol. 20080;26(2):271-8. PMID 18182667
- ↑ Dent et al.: Searching for a cure: Gene therapy for glioblastoma. Cancer Biol Ther 2008, 7(9) (elektronische Vorabpublikation) PMID 18708757.
- ↑ Maier-Hauff et al.: Intracranial thermotherapy using magnetic nanoparticles combined with external beam radiotherapy: results of a feasibility study on patients with glioblastoma multiforme. J Neurooncol 2007, 81, 53–60; PMID 16773216.
- ↑ Jordan et. al.: The effect of thermotherapy using magnetic nanoparticles on rat malignant glioma. J Neurooncol 2006, 78, 7–14; PMID 16314937.
- ↑ Krex et al.: Long-term survival with glioblastoma multiforme. Brain 2007, 130, 2596–2606; PMID 17785346.
Weblinks
Wikimedia Foundation.
Schlagen Sie auch in anderen Wörterbüchern nach:
glioblastoma multiforme — Tumor maligno de crecimiento rápido, pulposo o quístico, del cerebro o, en ocasiones, de la médula espinal. La lesión se disemina mediante proyecciones en forma de seudópodos. Diccionario Mosby Medicina, Enfermería y Ciencias de la Salud,… … Diccionario médico
Glioblastoma multiforme — DiseasesDB = 29448 MeshID = D005909 Glioblastoma multiforme (GBM) is the most common and most aggressive type of primary brain tumor, accounting for 52% of all primary brain tumor cases and 20% of all intracranial tumors. Despite being the most… … Wikipedia
glioblastoma multiforme — GBM. A fast growing type of central nervous system tumor that forms from glial (supportive) tissue of the brain and spinal cord and has cells that look very different from normal cells. Glioblastoma multiforme usually occurs in adults and affects … English dictionary of cancer terms
glioblastoma multiforme — Eng. Glioblastoma multiforme Tumor cerebral maligno con gran tendencia infiltrativa y rápido crecimiento. Aunque su localización más frecuente es la hemisférica puede asentar o afectar de forma secundaria al quiasma óptico, nervio óptico u órbita … Diccionario de oftalmología
glioblastoma multiforme — A glioma consisting chiefly of undifferentiated anaplastic cells of astrocytic origin that show marked nuclear pleomorphism, necrosis, and vascular endothelial proliferation; frequently, tumor … Medical dictionary
glioblastoma multiforme — noun see glioblastoma … New Collegiate Dictionary
glioblastoma multiforme — noun The most common and most aggressive type of primary brain tumor … Wiktionary
multiforme — see ERYTHEMA MULTIFORME, GLIOBLASTOMA MULTIFORME … Medical dictionary
glioblastoma — A fast growing type of central nervous system tumor that forms from glial (supportive) tissue of the brain and spinal cord and has cells that look very different from normal cells. Glioblastoma usually occurs in adults and affects the brain more… … English dictionary of cancer terms
glioblastoma — noun (plural mas; also glioblastomata) Etymology: New Latin, from glia glia + blast + oma Date: circa 1923 a malignant rapidly growing astrocytoma of the central nervous system and usually of a cerebral hemisphere called also glioblastoma… … New Collegiate Dictionary
 18+© Academic, 2000-2026
18+© Academic, 2000-2026- Kontaktieren Sie uns: Unterstützung, Werbung
Wörterbücher Export, schritte mit PHP, Joomla, Drupal, WordPress, MODx.
.jpg)