- Klassifikation nach Fredrickson
-
Klassifikation nach ICD-10 E78 Störungen des Lipoproteinstoffwechsels und sonstige Lipidämien E78.0 Reine Hypercholesterinämie E78.1 Reine Hypertriglyzeridämie E78.2 Gemischte Hyperlipidämie E78.3 Hyperchylomikronämie E78.6 Lipoproteinmangel ICD-10 online (WHO-Version 2006) Unter Hyperlipoproteinämie (HLP) oder Hyperlipidämie versteht man allgemein eine erhöhte Konzentration des Cholesterins, der Triglyceride, und der Lipoproteine mit Verschiebung des relativen Anteils der LDL- bzw. VLDL-Fraktion.
Man unterscheidet zwischen primären und sekundären Hyperlipoproteinämien. Primäre Hyperlipoproteinämien stellen eine eigene, meist genetisch bedingte Erkrankung dar, während sekundäre Hyperlipoproteinämien Folgeerscheinungen von anderen Grunderkrankungen sind.
Inhaltsverzeichnis
Primäre Hyperlipoproteinämien
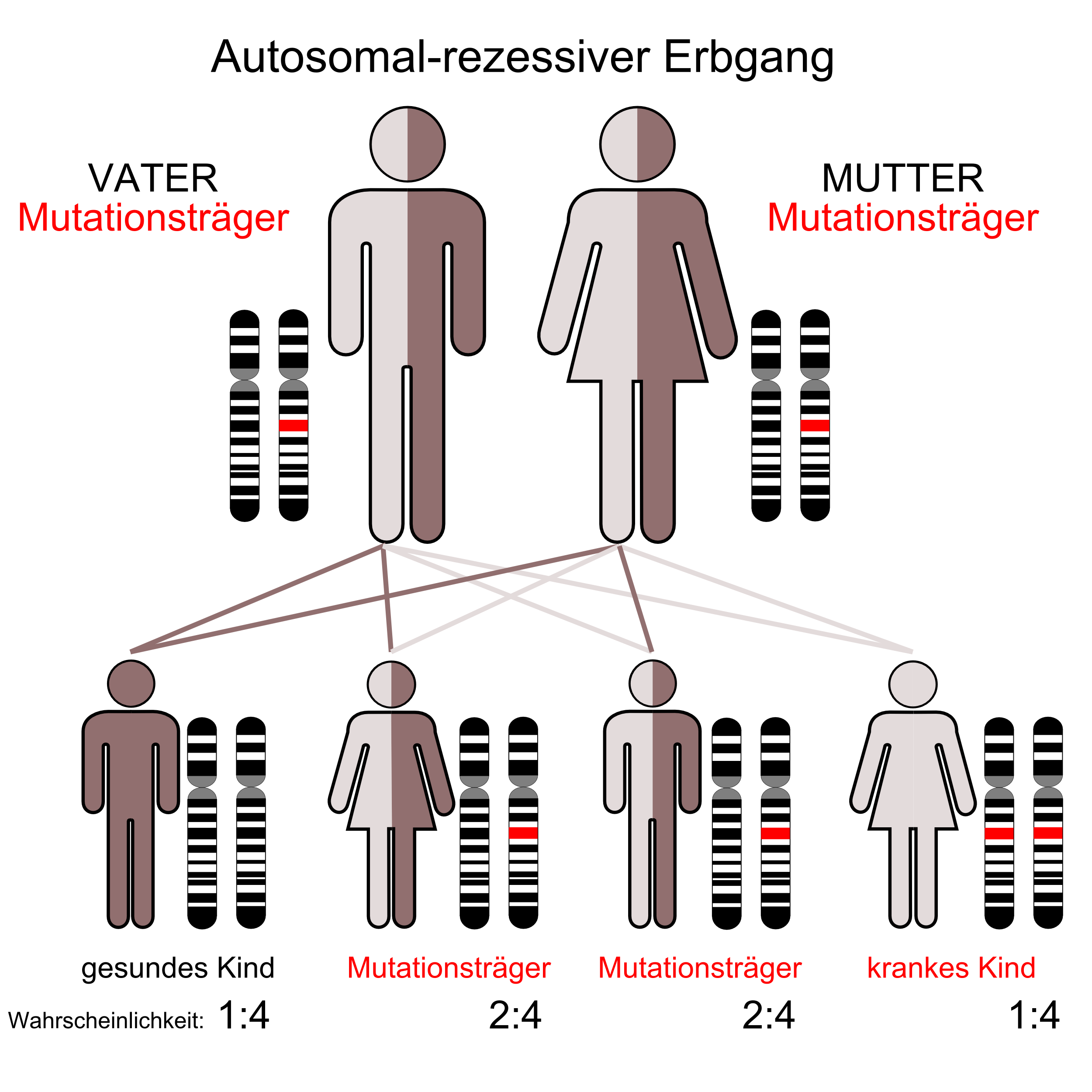
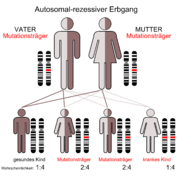 Der autosomal-rezessive Erbgang
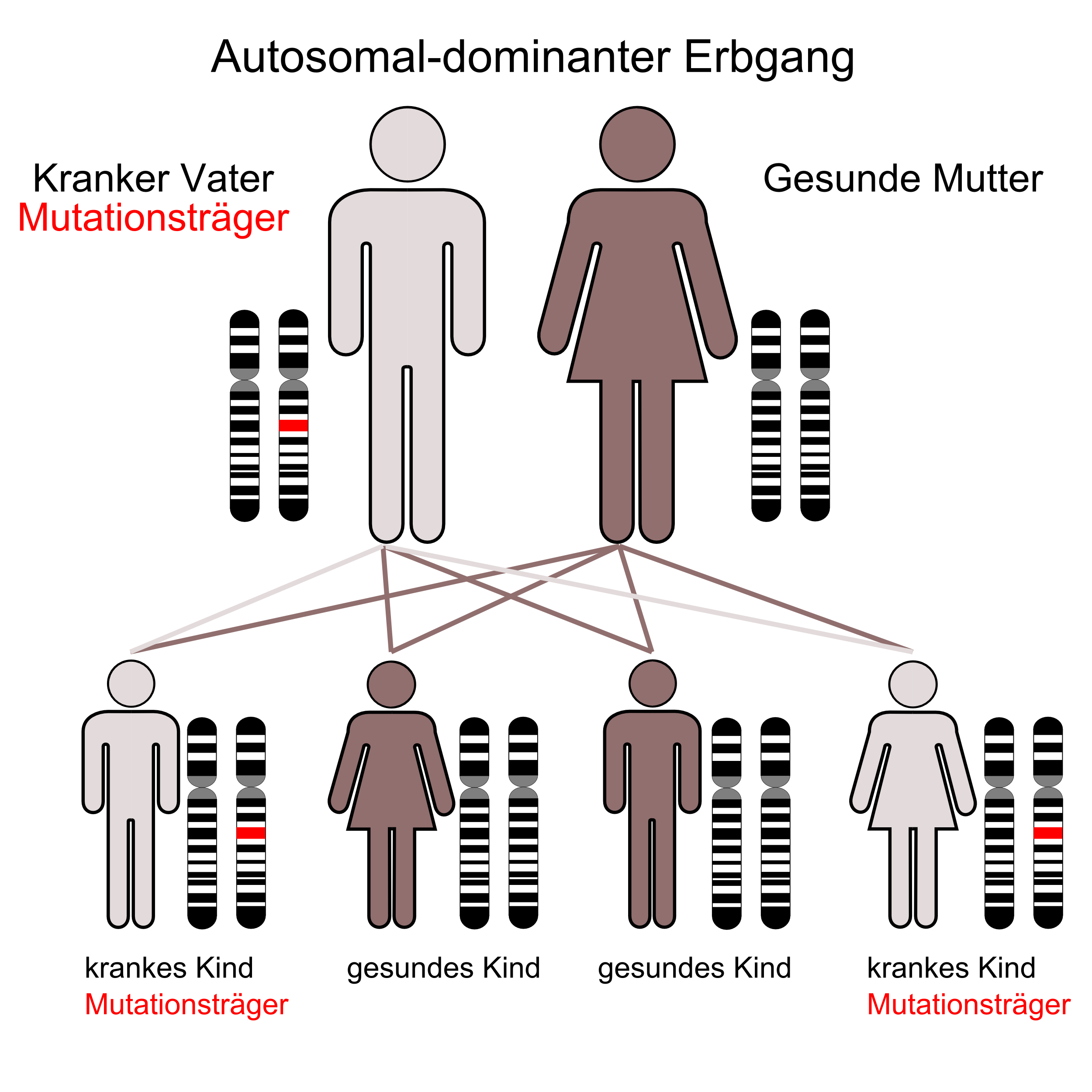
Der autosomal-rezessive Erbgang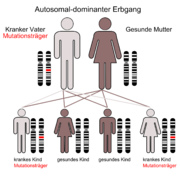 Der autosomal-dominante Erbgang
Der autosomal-dominante ErbgangDie Primären Hyperlipoproteinämien können anhand der Klassifikation nach Fredrickson unterschieden werden. Es werden die Werte für Gesamtcholesterin, LDL-Cholesterin und HDL-Cholesterin sowie die Triglyceride berücksichtigt. Die Färbung des Nüchternserums und das Ergebnis der Gelelektrophorese (Lipid-Elektrophorese) gehen ebenfalls in die Klassifikation ein.
HLP Typ 1 (Hypertriglyceridämie)
Selten; autosomal rezessiv vererbt; Defekt des posthepatischen Lipoproteinlipase-Systems; stark verzögerter Abbau der Chylomikronen; Es kommt zu einer pathologischen Ablagerung von Lipiden in Leber, Milz, und in der Haut (Xanthome). Bei der HLP Typ 1 besteht kein erhöhtes Risiko für Arteriosklerose.
Die HLP Typ 1 zeichnet sich im Labor durch normale Gesamtcholesterinwerte und erhöhte Triglyceride aus. LDL-Cholesterin und HDL-Cholesterin sind niedriger als die Referenzwerte. Das Nüchternserum ist unten klar und zeigt sich an der Oberfläche "aufrahmend". Bei der Gelelektrophorese ist die Bande der Chylomikronen sehr deutlich, die anderen Banden (beta, prä-beta und alpha) immer noch deutlich zu erkennen.
HLP Typ 2 (Hypercholesterinämie)
Häufig; autosomal dominant vererbt; Defekt des LDL-Rezeptors infolge einer Mutation des Rezeptorgens (auf Chromosom 19); Da LDL nicht bzw. nur gering in die Leber aufgenommen werden kann geht die Rückkopplungshemmung der Cholesterinsynthese verloren, was zu einer exzessiven VLDL Synthese der Leber führt. Das daraus gebildete LDL erhöht massiv den Serumcholesterinspiegel.
Bei HLP Typ 2a kommt es zu einer isolierten Erhöhung von LDL mit den oben beschriebenen Folgen, während bei Typ 2b zusätzlich auch TG leicht erhöht sind. Dies ist wahrscheinlich durch eine Überproduktion von ApoB bedingt, sodass die VLDL Bildung vermehrt ist. Patienten mit HLP Typ 2 bekommen schon im Kindesalter massive Arteriosklerose und Durchblutungsstörungen und erleiden früh Herzinfarkte.
Typ 2a zeichnet sich im Gegensatz zu Typ 1 durch erhöhte Gesamtcholesterinwerte und normale Triglyceridwerte aus. Das LDL-Cholesterin ist erhöht, das HDL-Cholesterin oft erniedrigt bis normal. Das Nüchternserum ist klar. Eine Gelelektrophorese zeigt die Banden alpha und prä-beta. Xanthome sind tendinös/tuberös.
Typ 2b, synonym für gemischte Hyperlipidämie, hat sowohl erhöhte Gesamtcholesterinwerte als auch erhöhte Triglyceridwerte. LDL-Cholestern ist erhöht, HDL-Cholesterin oft erniedrigt. In der Gelelektrophorese zeigen sich starke beta- und prä-beta-Banden, die alpha-Bande ist sichtbar. Das Nüchternserum ist leicht trüb. Xanthome sind tendinös und tuberös.
HLP Typ 3 (Remnant Hyperlipidämie, Broad-beta-Disease)
Sehr selten; autosomal dominant vererbt; Abnormes Apolipoprotein E führt zu unvollständigem Abbau der Chylomikronenremnants und der IDL, sodass deren Plasmakonzentrationen ansteigen. Folgen sind ein massiv erhöhtes Arterioskleroserisiko.
Typ 3 weist wie auch Typ 2b erhöhte Gesamtcholesterinwerte und Triglyceride im Labor auf. LDL-Cholesterin ist oft normal bis leicht erhöht. HDL-Cholesterin meist erniedrigt. In der Gelelektrophorese zeigt sich eine deutliche prä-beta Bande und eine leichtere alpha-Bande. Das Nüchternserum ist trüb.
HLP Typ 4 (Hypertriglyzeridämie)
Häufig; autosomal dominant vererbt; Es liegt eine Überproduktion endogener Triglyzeride sowie eine verminderte Verwertung von VLDL-Triglyzeriden vor, was zu einer VLDL-Erhöhung führt. Folgen sind Oberbauchkoliken (aufgrund von Pankreatitis), erhöhtes Arterioskleroserisiko, Übergewicht, Fettleber, Hyperurikämie (erhöhter Harnsäurespiegel, was zu Gicht führt) uvm.
Typ 4 zeigt im Labor ein normales bis leicht erhöhtes Gesamtcholesterin und deutlich erhöhte Triglyceridwerte. LDL-Cholesterin ist meist normal, HDL-Cholesterin oft erniedrigt. Das Nüchternserum ist trüb. Die Gelelektrophorese weist eine deutliche prä-beta-Bande und eine schwächere beta- und alpha-Bande auf.
HLP Typ 5 (Kombinierte Hyperlipidämie, endogen-exogene-Hypertriglyceridämie)
Die Pathogenese der HLP Typ 5 ist unklar. Die Folgen sind Übergewicht, Fettleber, Hepatosplenomegalie, Oberbauchkoliken und Xanthome der Haut. Das Arterioskleroserisiko ist nicht erhöht.
Typ 5 zeigt im Labor normale bis erhöhte Gesamtcholesterinwerte und erhöhte Triglyceridwerte. LDL-Cholesterin ist meist erhöht, HDL-Cholesterin oft erniedrigt. Das Nüchternserum ist trüb und aufrahmend. Die Gelelektrophorese zeigt deutliche prä-beta und Chylomikronenbanden und schwächere beta- und alpha-Banden.
Sekundäre Hyperlipoproteinämien
Die Ursachen von Sekundären Hyperlipoproteinämien liegen in folgenden Grunderkrankungen:
- Alkoholismus
- Überernährung
- Fehlernährung
- Nephrotisches Syndrom
- Pankreatitis
- Lebererkrankungen
- Cholestase
- Diabetes Mellitus
- Hyperurikämie
- Hypothyreose
- Akromegalie
- Glykogenosen
- Hyperkalzämie
In der Regel erfolgt die Heilung der sekundären Hyperlipoproteinämien durch die Behandlung der jeweiligen Grundkrankheit. Auch durch die Einnahme von Ovulationshemmern und durch eine Schwangerschaft kann vorübergehend eine Hyperlipoproteinämie entstehen.
Quellen
- Regine Witkowski et.al.: Lexikon der Syndrome und Fehlbildungen. Ursachen, Genetik und Risiken 7. Auflage, Seiten 579ff., Springer Verlag Berlin 2004, ISBN 978-3-540-44305-6
- Prof. Dr. P.B. Luppa et.al.: Skript Praktikum der Klinischen Chemie und Hämatologie 6. Auflage, Institut für Klinische Chemie und Pathobiochemie Technische Universität München 2006

Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.