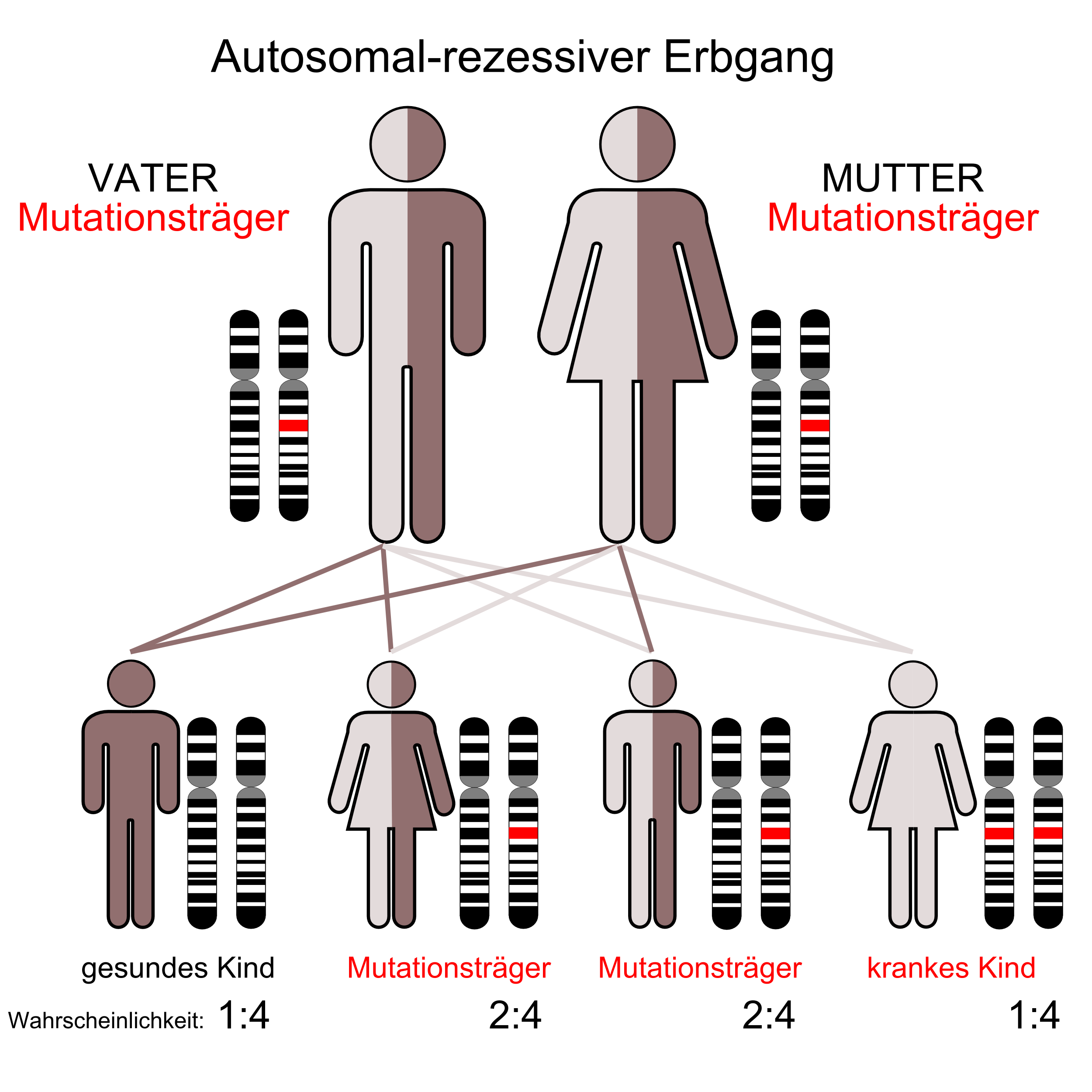
- Familiäre Hypoalphalipoproteinämie
-
Klassifikation nach ICD-10 E78.6 Lipoproteinmangel
Tangier-KrankheitE75.6 systematisierte Lipidablagerungserkrankung ICD-10 online (WHO-Version 2006) Die Tangier-Krankheit ist eine seltene Erbkrankheit des Fettstoffwechsels. Bei dieser Erkrankung ist die Freisetzung von Cholesterin aus der Zelle gestört, wodurch es einerseits zu einer verminderten Bildung von High Density Lipoproteinen sowie andererseits zu einer vermehrten Cholesterinspeicherung insbesondere in Zellen des retikulohistiozytären Systems kommt. Leitsymptom sind gelb-orange Flecken auf vergrößerten Rachenmandeln. Zudem können Erkrankungen der Nerven, Vergrößerungen von Bauchorganen und gehäuft Arteriosklerose auftreten. Die Prognose ist insgesamt günstig, eine kausale Therapie derzeit nicht möglich.
Der Begriff „Tangier-Krankheit“ wird in der Literatur gleichbedeutend mit „An-α-Lipoproteinämie“[1], „Familiärer HDL-Mangel“[2] und „familiärer Hypoalphalipoproteinämie“[3] verwendet.
Inhaltsverzeichnis
Geschichtliche Aspekte und Verbreitung
Die Erkrankung wurde vom Erstbeschreiber[5] Donald S. Fredrickson 1961 nach der isoliert vor der Küste Virginias liegenden Insel Tangier benannt, da die beiden ersten beschriebenen Patienten von dort stammten.[6][7]
Es handelt sich um eine seltene Erkrankung. Bis 1978 sind weltweit 25 Fälle, bis 1987 35 Fälle (davon 21 mit begleitenden neurologischen Symptomen) und bis 2005 über 100 Patienten berichtet worden.[4]
Ursache und Krankheitsentstehung
Die Tangier-Krankheit wird autosomal-rezessiv vererbt.[4] Der zugrunde liegende Gendefekt ist auf dem langen Arm des Chromosom 9 lokalisiert (9q31) und betrifft das ABCA 1-Gen. Letzteres kodiert für ein Transportprotein, das für die Auschleusung von Cholesterin aus der Zelle verantwortlich ist.[8]
Durch den Gendefekt ist die Ausschleusung von Cholesterin aus der Zelle gestört, das so nicht zusammen mit Apolipoprotein A-I zur Bildung von High Density Lipoprotein im Blutserum beitragen kann. Hierdurch kommt es zu einem HDL-Mangel mit Serumkonzentrationen, die im Vergleich zu Normalpersonen um das 100–200fache erniedrigt sind, während sich in der Zelle Cholesterinester ansammeln.[9]
Diagnose
Als Leitsymptom finden sich bereits im Kindesalter[5] hypertrophe, gelb- bis orangegefärbte Rachenmandeln. Fredrickson dokumentierte diese Veränderung bereits in seiner Erstbeschreibung photographisch.[10] Wurden die Tonsillen entfernt, finden sich als zuverlässige Befunde kleine (1-2 mm Durchmesser) Flecken in der Schleimhaut des Afters.[11] Wegweisend für die Diagnose sind, neben den typisch veränderten Rachenmandeln zudem die Spiegel von Gesamtcholesterin, Triglyceriden, HDL-Cholesterin und Apolipoprotein A-I im Blut. Die α- und Prä-β-Bande in der Lipoprotein-Elektrophorese fehlt. Der Cholesterinspiegel im Blut liegt unter 100 mg/dl (meist unter 25 mg/dl)[5], das HDL-Cholesterin fehlt, oder das wenige, das in einigen Fällen noch vorhandenen ist, ist fehlstrukturiert (wie auch LDL und VLDL). Nicht davon betroffen sind die Triglyceride, die sogar erhöht sein können.[3][4] Erniedrigt ist meist ebenfalls das Apolipoprotein A-II.[11]
Der klinische Verdacht auf das Vorliegen einer Tangier-Krankheit kann durch eine humangenetische Untersuchung bestätigt werden.[11] Eine Biopsie des Nerven ist zur Diagnosestellung üblicherweise nicht erforderlich.[12]
Klinisches Bild
Neben den bereits beschriebenen Vergrößerung und Veränderungen der Rachenmandeln[5] können Muskelschwäche und Nervensymptome, insbesondere Sensibilitätsstörungen an den Beinen, sowie eine Vergrößerung von Leber und Milz auftreten.[5] Auch ist in einem Einzelfall eine massive Vergrößerung der Bauchspeicheldrüse beschrieben.[13]
Die Abwesenheit des HDL- Cholesterins begünstigt die Entstehung einer Arteriosklerose. Der Verlauf ist jedoch individuell unterschiedlich, so dass eine koronare Herzkrankheit bei manchen Patienten erst in fortgeschrittenem Lebensalter auftritt.[4][14][15] Hornhauttrübungen und Blutbildveränderungen (z. B. hämolytische Anämie, Mangel an Blutplättchen) wurden ebenfalls beschrieben.[11]
Die durch Erkrankung des peripheren Nervensystems entstehende neurologische Symptomatik kann sich in drei unterschiedlichen Formen bemerkbar machen. Zum einen als distale sensomotorische Neuropathie mit Gefühls- und Bewegungsstörung der Beine und Arme, des weiteren als einzelne Nerven wiederkehrend betreffende Neuropathie (rezidivierende Mononeuropathie) sowie als ein im Erwachsenenalter beginnendes an eine Syringomyelie erinnerndes (pseudosyringomyelitisches) Syndrom mit Schwäche der Gesichtsmuskulatur und der kleinen Muskeln der Hand, fehlenden oder schwachen Muskeleigenreflexen, Schmerz und Temperaturempfindungs- und später auch allgemeine Gefühlsstörungen an Rumpf und stammnahen Extremitäten.
Die Nervenleitgeschwindigkeit ist bei den beiden erstgenannten Verlaufsformen normal.[12] Inwieweit sich diese Krankheitsformen nicht nur feingeweblich sondern auch genetisch unterscheiden, ist bislang nicht abschließend geklärt[4][11][12], aber Gegenstand der Forschung.[16]
Pathologie
Bei der Tangier-Krankheit lagern sich Cholesterinester in den Makrophagen[5] verschiedener Gewebe ab (z. B. peripheres und zentrales Nervensystem, Milz, Lymphknoten, Knochenmark, retikuloendotheliales System, Thymus, Mastdarmschleimhaut und Haut). Diese Ablagerungen sind ursächlich für die klinisch auffällige typische Gelb-Orangefärbung der Tonsillen.[4][5] Feingeweblich sind in den befallenen Geweben die fettbeladenen Makrophagen als Schaumzellen nachweisbar.[3][11][17]
In der Nervenbiopsie findet sich bei Patienten mit der seltenen syringomyelie-ähnlichen Verlaufsform eine teils ausgeprägte Reduktion myelinisierter und unmyelinsierter Axone bei Nachweis einer Vakuolisierung von Schwann-Zellen und endoneuralen Makrophagen.[18] Bei den (mono)-neuropathischen Formen können unter Umständen lediglich unspezifische Zeichen einer De- und Remyelinisation nachweisbar sein.[19]
Behandlung und Prognose
Eine kausale Behandlung der Erkrankung ist bislang nicht möglich, gentechnische Ansätze sind jedoch vorstellbar.[3][11] Allgemein wird eine fettarme Ernährung empfohlen.[20]
Die Prognose wird als relativ günstig eingeschätzt.[21] Im Erwachsenenalter besteht jedoch ein erhöhtes Risiko für die Entwicklung von Gefäßerkrankungen,[4][22][14] wobei eine valide Risikoeinschätzung auch wegen der niedrigen Zahl bekannter Fälle nicht möglich ist.
Einzelnachweise
- ↑ Roche Lexikon Medizin, Urban & Fischer, 2003, ISBN 3437151509; hier online
- ↑ Mayatepek E.: Pädiatrie mit Studentconsult-zugang, Urban&FischerVerlag, 2007, S.239, ISBN 3437435604; hier online
- ↑ a b c d Baenkler H.-W.: Innere Medizin, Thieme Verlag, 2001, S. 972, ISBN 3131287519; hier online
- ↑ a b c d e f g h Schröder J.-M., e.a.: Spezielle pathologische Anatomie, Springer, 1999, S. 367 ff., ISBN 354065612X; hier online
- ↑ a b c d e f g h Guder G., e.a.: Das Laborbuch für Klinik und Praxis, Urban&FischerVerlag, 2005, S. 252 ff., ISBN 3437233408; hier online
- ↑ Fredrickson et al. Tangier disease. Ann Int Med. 1961;55:1016.
- ↑ Remaley et al.: Human ATP-binding cassette transporter 1 (ABC1): genomic organization and identification of the genetic defect in the original Tangier disease kindred. Proc Natl Acad Sci U S A. 1999;96(22):12685-90. PMID 10535983 Volltext
- ↑ Brooks-Wilson et al.:Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet. 1999;22(4):336-45. PMID 10431236
- ↑ Young & Fielding: The ABCs of cholesterol efflux. Nat Genet. 1999;22(4):316-8. PMID 10431227
- ↑ a b profiles.nlm.nih.gov: Photograph of tonsils of a patient with Tangier Disease; hier online; zuletzt eingesehen am 18. Dez. 2008
- ↑ a b c d e f g Ganten D.: Handbuch der molekularen Medizin, Springer, S. 334ff, ISBN 3540655298; hier online
- ↑ a b c Berlit P.: Klinische Neurologie, Springer, 1999, S.197ff. ,ISBN 3540652817; hier online
- ↑ Sperti C., e.a.: Abdominal localization of Tangier disease mimicking a pancreatic neoplasm. In: Eur J Gastroenterol Hepatol., 10/2008, S. 1028-31; hier online
- ↑ a b Serfaty-Lacrosniere C, e.a.: Homozygous Tangier disease and cardiovascular disease. In: Atherosclerosis. 1994 May;107(1):85-98.Links hier online
- ↑ Kolovou G., e.a.: Postprandial hypertriglyceridaemia in patients with Tangier disease. In: J Clin Pathol. 2003 December; 56(12): 937–941, hier online
- ↑ Züchner et al.: A novel nonsense mutation in the ABC1 gene causes a severe syringomyelia-like phenotype of Tangier disease. Brain. 2003;126(Pt 4):920-7. PMID 12615648
- ↑ Siegenthaler W., e.a.: Klinische Pathophysiologie, Thieme Verlag, 2006, S. 158, ISBN 3134496097; hier online
- ↑ Gibbels et al.: Severe polyneuropathy in Tangier disease mimicking syringomyelia or leprosy. Clinical, biochemical, electrophysiological, and morphological evaluation, including electron microscopy of nerve, muscle, and skin biopsies. J Neurol. 1985;232(5):283-94. PMID 2997405
- ↑ Pollock et al.:Peripheral neuropathy in Tangier disease. Brain. 1983;106 ( Pt 4):911-28. PMID 6317140
- ↑ Reuter P.: Springer Lexikon Medizin, Springer, 2004, ISBN 3540204121; hier online
- ↑ Begemann H., e.a.: Handbuch der inneren Medizin, Springer, 1968, S. 268, ISBN 0387063552; hier online
- ↑ Schumacher H. R., e.a.: Handbook of Hematologic Pathology, Informa Health Care, 2000, S.161-2 , ISBN 0824701704, hier online
Wikimedia Foundation.
Schlagen Sie auch in anderen Wörterbüchern nach:
Liste der Endokrinen, Ernährungs- und Stoffwechselkrankheiten nach ICD-10 — Die Internationale statistische Klassifikation der Krankheiten und verwandter Gesundheitsprobleme (ICD 10 in seiner letzten international gültigen Version von 2006) klassifiziert im Kapitel IV die Endokrinen, Ernährungs und… … Deutsch Wikipedia
 18+© Academic, 2000-2026
18+© Academic, 2000-2026- Kontaktieren Sie uns: Unterstützung, Werbung
Wörterbücher Export, schritte mit PHP, Joomla, Drupal, WordPress, MODx.