- Lynch-Syndrom
-
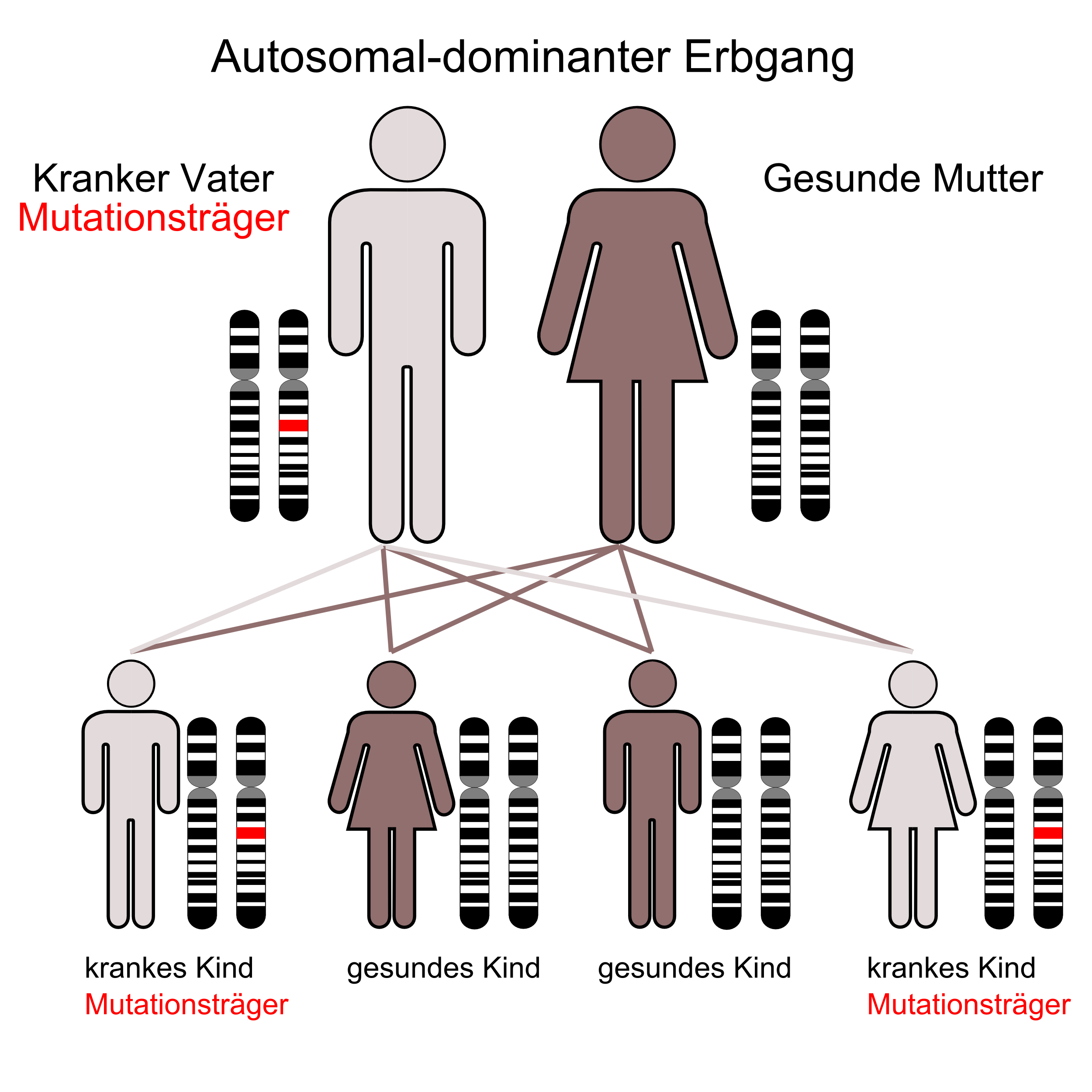
Klassifikation nach ICD-10 C18.9 Bösartige Neubildung: Kolon, nicht näher bezeichnet Z80.0 Bösartige Neubildung der Verdauungsorgane in der Familienanamnese ICD-10 online (WHO-Version 2006) 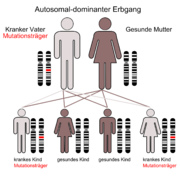 Der autosomal-dominante Erbgang
Der autosomal-dominante ErbgangDas hereditäre non-polypöse Kolonkarzinom (HNPCC) oder Lynch-Syndrom ist eine erbliche Darmkrebsform ohne Polyposis, d. h. ohne Auftreten von vielen Polypen im Darm, die autosomal-dominant vererbt wird.
Inhaltsverzeichnis
Epidemiologie
Das hereditäre Dickdarm-Karzinom ohne Polyposis (HNPCC) ist die häufigste erbliche Darmkrebsform und betrifft etwa 5 % der Darmkrebsfälle. Bei etwa 75 % der Genträger tritt ein Kolonkarzinom auf. Das mittlere Alter bei Diagnose liegt bei 45 Jahren.
Kinder von Patienten haben entsprechend dem Erbgang eine Wahrscheinlichkeit von 50 %, Genträger zu sein.
Pathophysiologie und Pathologie
Meist kommt es beim hereditären Dickdarm-Karzinom ohne Polyposis (HNPCC) zum Auftreten mehrerer bösartiger Darmtumoren entweder synchron (gleichzeitig) oder metachron (zeitlich versetzt). Die Tumoren sind in mehr als zwei Drittel der Fälle im proximalen Colon (rechtsseitiger Grimmdarm und Blinddarm) lokalisiert. Neben dem Auftreten von Darmkrebs kommt es bei Frauen überdurchschnittlich häufig zum Auftreten von Endometriumkarzinomen (Gebärmutterkrebs) (kumulatives Risiko bis zum 70. Lebensjahr etwa 20 %, nach anderen Quellen 40 % - 60 %). Des weiteren findet sich ein familiär gehäuftes Auftreten von Magenkrebs (6 %), Karzinomen des Dünndarms (1 %), Leberkrebs und Gallengangskrebs (4 %), Krebs der ableitenden Harnwege (2 %), Eierstockkrebs (1 – 3 %) sowie Bauchspeicheldrüsenkrebs (2 %), Hautkrebs (2 %) und Hirntumoren (3 %, meist Glioblastoma), sowie Talgdrüsenadenome und Keratoakanthome (bei Muir-Torre-Syndrom).
Molekularbiologie des hereditären non-polypösen Karzinoms
Charakteristischerweise werden mittels molekularbiologischer Methoden bei Betroffenen Veränderungen der Mikrosatellitenmarker nachgewiesen. Bei Patienten mit hereditären non-polypösen Karzinom findet man innerhalb eines betroffenen Individuums einer Sequenzlängendifferenz zwischen Tumor und gesundem Gewebe als Hinweis auf eine fehlerhafte DNA-Replikation. Man bezeichnet dieses Phänomen als Mikrosatelliteninstabilität.
Mittlerweile sind 6 Gene bekannt, deren Keimbahnmutation zum Auftreten eines hereditären non-polypösen Karzinoms führen kann: hMSH2 (31%), hMLH1 (33%), hPMS1 (2%), hPMS2 (4%), hMSH6 und hMLH3. Diese Gene gehören zur Gruppe der so genannten DNA-Mismatch-Reparaturgene, deren Aufgabe es ist, eventuelle Fehler bei der Replikation der DNA im Rahmen der Zellteilung zu erkennen und zu beseitigen.
Diagnosekriterien
Amsterdam I-Kriterien
Alle Kriterien müssen erfüllt sein
- mindestens drei Familienangehörige mit histologisch gesichertem kolorektalem Karzinom
- einer davon Verwandter ersten Grades der beiden anderen
- Erkrankungen in mindestens zwei aufeinanderfolgenden Generationen
- mindestens ein Patient mit der Diagnose des kolorektalen Karzinoms vor dem 50. Lebensjahr
- Ausschluss einer FAP (Familiäre adenomatöse Polyposis)
Amsterdam II-Kriterien
Alle Kriterien müssen erfüllt sein.
- mindestens drei Familienangehörige mit HNPCC-assoziiertem Karzinom (Kolon/ Rektum, Endometrium, Dünndarm, Nierenbecken/Ureter)
- einer davon Verwandter ersten Grades der beiden anderen
- Erkrankungen in mindestens zwei aufeinanderfolgenden Generationen
- mindestens ein Patient mit der Diagnose eines Karzinoms vor dem 50. Lebensjahr
- Ausschluss einer FAP (Familiäre adenomatöse Polyposis)
Revidierte Bethesda-Kriterien
Mindestens ein Kriterium muss erfüllt sein.
Tumoren von Patienten sollten auf das Vorliegen einer Mikrosatelliten-Instabilität in folgenden Fällen untersucht werden:
- Patienten mit kolorektalem Karzinom vor dem 50. Lebensjahr.
- Patienten mit synchronen oder metachronen kolorektalen Karzinomen oder anderen HNPCC-assoziierten Tumoren)*, unabhängig vom Alter.
- Patienten mit kolorektalem Karzinom mit MSI-H Histologie)** vor dem 60. Lebensjahr.
- Patient mit kolorektalem Karzinom (unabhängig vom Alter), der einen Verwandten 1. Grades mit einem kolorektalen Karzinom oder einem HNPCC-assoziierten Tumor vor dem 50. Lebensjahr hat.
- Patient mit kolorektalem Karzinom (unabhängig vom Alter), der mindestens zwei Verwandte 1. oder 2. Grades hat, bei denen ein kolorektales Karzinom oder ein HNPCC-assoziierter Tumor (unabhängig vom Alter) diagnostiziert wurde.
)*zu den HNPCC-assoziierten Tumoren gehören Tumoren in: Kolorektum, Endometrium, Magen, Ovarien, Pankreas, Ureter oder Nierenbecken, Gallengang, Dünndarm und Gehirn (meist Glioblastome wie bei Turcot-Syndrom) sowie Talgdrüsenadenome und Keratoakanthome (bei Muir-Torre-Syndrom)
)** Vorliegen von Tumor-infiltrierenden Lymphozyten, Crohn-ähnlicher lymphozytärer Reaktion, muzinöser/Siegelring-Differenzierung, oder medullärem Wachstumsmuster
Vererbungsmuster
Es gibt zwei Vererbungsmuster.
Lynch-Syndrom Typ I
In diesen Familien tritt von Generation zu Generation fast ausschließlich Dickdarmkrebs auf.
Lynch-Syndrom Typ II
In diesen Familien wird neben Darmkrebs vor allem auch Gebärmutter- und Eierstockkrebs festgestellt, sowie Krebs an Nieren, Leber, Magen, Dünndarm.
Früherkennungsprogramm zur Vor- und Nachsorge
Vom Verbundprojekt „Familiärer Dickdarmkrebs“ wird ein lebenslängliches Früherkennungsprogramm für HNPCC-Patienten empfohlen:
Einmal jährlich zusätzlich zu den sonstigen Nachsorgen wie Blutuntersuchungen, okkultes Blut im Stuhl, Brust:
- Körperliche Untersuchung
- Abdomensonographie
- Koloskopie
- Gynäkologische Untersuchung auf Endometrium- und Ovarialcarzinom (inkl. transvaginaler Sonographie)
- Urinzytologie
- Ösophago-Gastro-Duodenoskopie (nur bei familiär gehäuften Magenkarzinomen)
Für Familienangehörige in Erblinie gelten die gleichen jährlichen Untersuchungsempfehlungen zur Vorsorge, es sei denn auf molekulargenetischer Ebene konnte der Nachweis erbracht werden, dass der Gen-Defekt nicht geerbt wurde. Der Beginn der Untersuchungen wird ab dem 25. Lebensjahr empfohlen, spätestens aber 5 Jahre vor dem zuerst in der Familie aufgetretenen Fall (Bsp.: jüngste Betroffene in der Familie war bei Auftreten der Erkrankung 25 J. alt >>> Vorsorgeuntersuchungen sollten für alle Familienmitglieder spätestens mit dem 20. Lebensjahr beginnen).
Siehe auch
Weblinks
- Verbundprojekt der Deutschen Krebshilfe "Familiärer Darmkrebs"
- [1] Infos zum Genetischen Beratungsgespräch des Medizinisch Genetischen Beratungszentrums in München
- Vererbbare Tumordispositionserkrankungen - Homepage der Arbeitsgruppe “Hereditäre Tumordisposition und molekulare Onkologie” am Klinikum rechts der Isar der Technischen Universität München
- Felix-Burda-Stiftung

Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.