- Fragiles-X-Syndrom
-
Klassifikation nach ICD-10 Q99.2 Fragiles X-Chromosom Syndrom des fragilen X-Chromosoms
ICD-10 online (WHO-Version 2011) Das Fragiles-X-Syndrom (FXS) ist eine der häufigsten Ursachen erblicher kognitiver Behinderung des Menschen. Ursache hierfür ist eine genetische Veränderung auf dem X-Chromosom, eine Mutation des Gens FMR1 (fragile X mental retardation 1). Das Syndrom wird nach seinen Erstbeschreibern auch als Martin-Bell-Syndrom (MBS) oder Marker-X-Syndrom sowie in der abgekürzten Form als fra(X)-Syndrom bezeichnet. Die Behinderung kann in ihrer Schwere stark variieren und von leichten Lernschwierigkeiten bis zu extremer kognitiver Beeinträchtigung reichen. Der Name leitet sich aus der Beobachtung von Zellkulturen betroffener Menschen ab: Unter entsprechenden Kulturbedingungen kann am X-Chromosom in einem Teil der Zellen eine Bruchstelle, der sogenannte fragile Bereich, nachgewiesen werden.
Inhaltsverzeichnis
Symptome
Vom Fragiles-X-Syndrom können sowohl Männer als auch Frauen betroffen sein. Das Leitsymptom ist eine unterschiedlich stark ausgeprägte Intelligenzminderung, deren Schwere von Lernproblemen bis hin zu schwergradiger kognitiver Beeinträchtigung reichen kann und mit Sprachstörungen und Aufmerksamkeitsdefiziten einhergeht.
Bei Kindern sind bei etwa 12 % der Betroffenen autistische Verhaltensweisen ausgeprägt, beinahe 20 % der Kinder bekommen Krampfbeschwerden (Epilepsie).
Bei Frauen sind die Symptome häufig milder ausgeprägt, was auf die zufällige Inaktivierung eines der beiden X-Chromosomen in weiblichen Zellen zurückzuführen ist.
Bei 80 % der betroffenen Männer findet sich eine Hodenvergrößerung, die schon vor der Pubertät auftreten kann. Weitere körperliche Symptome können eine vorspringende Stirn, abstehende und große Ohren und ein hervorstehendes Kinn bei gleichzeitig bestehendem schmalen Gesicht sein.
Verbreitung
Die Häufigkeit des Fragiles-X-Syndroms wird in der Literatur sehr breit angegeben, da in vielen Studien auch unterschiedliche Bemessungsgrundlagen für eine Vollmutation angelegt wurden.
Im Schnitt beträgt die Häufigkeit 1:1.200 bei Männern und 1:2.500 bei Frauen. Damit stellt diese Besonderheit nach dem Down-Syndrom (Trisomie 21) die häufigste Form von genetisch bedingter kognitiver Behinderung dar.
Diagnose
Bis zur Entdeckung des zugrundeliegenden Gens im Jahre 1991 war der Nachweis einer Lücke im X-Chromosom in Zellkulturen das einzige, allerdings recht unzuverlässige Verfahren, da in benachbarten Genbereichen ebenfalls fragile Stellen auftreten. Die Diagnostik erfolgt heute über molekulargenetische Analysemethoden aus einer Blutprobe mittels Polymerase-Kettenreaktion (PCR) und Southern Blot. Bleibt trotz negativer Befunde ein Verdacht, kann mittels immunohistochemischer Diagnostik mit monoklonalen Antikörpern direkt die FMR-Proteinkonzentration bestimmt werden. Für Feten mit erhöhtem Risiko kann pränatal entweder eine Amniozentese in der 16.–18. Schwangerschaftswoche (SSW) oder eine Chorionzottenbiopsie in der 10.–12. SSW durchgeführt werden.
Differenzialdiagnose
Da die Symptome in früher Kindheit oft unspezifisch sind und Entwicklungsverzögerungen für viele Menschen in Betracht kommen, ist die Differenzialdiagnose vergleichsweise schwierig.
Insbesondere in Betracht kommen das Sotos-Syndrom, das Prader-Willi-Syndrom, Autismus und die Aufmerksamkeitsdefizit-/Hyperaktivitätsstörung (ADS), mit der das Fragiles-X-Syndrom anscheinend mehrere auslösende Gene teilt.
Bei Kindern mit Sprech-, Sprachverzögerung und motorischen Defiziten sollte deshalb ein Test auf das Fragiles-X-Syndrom in Betracht gezogen werden, insbesondere bei entsprechender Familienanamnese.
Geschichtliches
Das Fragiles-X-Syndrom wurde 1943 erstmalig durch James Purdon Martin (1893–1984) und Julia Bell anhand einer Familie mit elf kognitiv zurückgebliebenen Männern unter wissenschaftlichen Gesichtspunkten beschrieben. Schon hier wurde ein X-chromosomaler Erbgang angenommen.
Erst 1969 konnte dies durch Herbert Lubs an einer vierköpfigen Familie mit zwei betroffenen Männern und zwei nicht betroffenen Frauen nachgewiesen werden. In seinen Zellkulturen beobachtete er ein Zusammenziehen des längeren Armes (q-Arm) in X-Chromosomen. Eine derartige Mutation konnte später auch in der ersten Familie nachgewiesen werden.
Die Entdeckung geriet zunächst in Vergessenheit, bis Grant Sutherland durch Zufall herausfand, dass der entsprechende Nachweis nur in einem Folsäure-freien Kulturmedium nachvollziehbar ist: Bei seinem Umzug von Melbourne nach Adelaide, wo ein anderes Kulturmedium eingesetzt wurde, welches bessere Chromosomenfärbungen ermöglichte, konnten seine Ergebnisse zunächst nicht wiederholt werden, bis er wieder das vorherige Kulturmedium einsetzte.
Bei der Vererbung des Fragiles-X-Syndroms war lange Zeit ungeklärt, warum es nicht immer mit anderen X-chromosomal gebundenen Erbgängen übereinstimmte. Insbesondere wurden auch heterozygote Überträgerinnen festgestellt, die eigentlich symptomfrei bleiben sollten. Dieses Phänomen veranlasste 1985 Stephanie Sherman und ihre Mitarbeiter zu einer genaueren Untersuchung der Stammbäume. Dabei stellten sie fest, dass Töchter eines nicht betroffenen Überträgers eine höhere Wahrscheinlichkeit besaßen, betroffene Nachkommen zu erhalten. Daraus schlussfolgerten sie, dass die Mutation in zwei Schritten erfolgen müsste, die im ersten Schritt noch symptomfrei bleibt und im zweiten nur bei der Übertragung von Frauen auf ihre Nachkommen erfolgt. Diese Beobachtungen sind in der Medizin seitdem als Sherman-Paradoxon bekannt.
Das die Erkrankung verursachende mutierte Gen wurde 1991 von mehreren Forschern gemeinsam entdeckt und das Syndrom in die Gruppe der Trinukleotiderkrankungen eingeordnet (Verkerk et al., 1991).
Genetische Ursache
 Position des Gens FMR1 auf dem X-Chromosom.
Position des Gens FMR1 auf dem X-Chromosom.
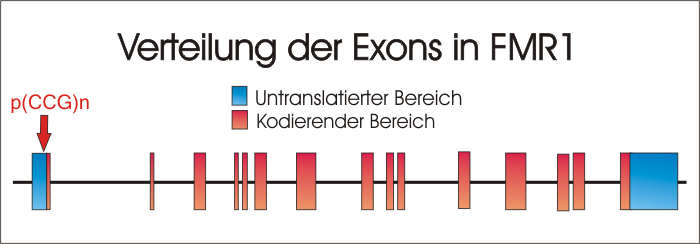
Genetische Ursache ist eine Veränderung des 38 kbp umfassenden Gens FMR1 ( Fragile X linked Mental Retardation type 1 ) in der Bande Xq27.3 des X-Chromosoms (oder bei der wesentlich selteneren Variante FraX-E[1] eine Veränderung des an Position Xq28 lokalisierten Gens FMR2). Das Gen FMR1 besteht aus 17 Exons und enthält eine sich wiederholende Sequenz aus CGG-Trinukleotiden. Der Normalbereich dieser Basentripletts pro Allel beträgt 6 bis 44 Wiederholungen, die in der Regel durch 2 AGG Tripletts an Position 9/10 oder 19/20 unterbrochen werden. Am häufigsten kommen 29 bis 30 Tripletts in der Normalbevölkerung vor.
 Die Position der verlängerten Basentripletts im FMR1 Gen sind mit einem roten Pfeil markiert.
Die Position der verlängerten Basentripletts im FMR1 Gen sind mit einem roten Pfeil markiert.Bei Menschen mit dem Fragiles-X-Syndrom treten zwei Arten von Mutationen in diesem Genbereich auf, die durch Verlängerung der CGG-Tripletts zustande kommen (Trinukleotidrepeat-Erkrankung). 59 bis 200 Wiederholungen werden als Prämutation bezeichnet und können im höheren Lebensalter mit einem eigenen Krankheitsbild, dem Fragiles-X-assoziierten Tremor-/Ataxie-Syndrom (FXTAS) einhergehen. Die Prämutation stellt eine Vorstufe der krankheitsverursachenden Vollmutation dar, die ab 200 oder mehr Wiederholungen gegeben ist. Dies führt zu einer Methylierung (einer Anlagerung von Methyl-Gruppen) des entsprechenden DNA-Abschnittes und damit zu einer Stilllegung der Genexpression des FMRP1-Proteins. Die Funktion dieses Proteins ist derzeit Gegenstand intensiver Forschung, wahrscheinlich ist es ein Schlüsselprotein bei der Herstellung weiterer Proteine, deren Fehlen zur Atrophie von Hirnzellen führt (Reiss et al., 1991). Die Methylierung führt auch zu dem Auflockern im betroffenen Bereich der Struktur des Chromosoms und damit zu dem typischen Bild des fragilen Bereiches.
Eine Anzahl von 45 bis 58 Wiederholungen der CGG-Tripletts wird als „graue Zone“ bezeichnet, bei der die Allele in der Regel stabil auf die Nachkommen übertragen werden. Da fast 2 % der Normalbevölkerung ein solches FMR1-Allel besitzen, ist dieser Bereich für die genetische Beratung prognostisch schwierig.
Die bisher kleinste bekannte Prämutation, bei der in einer Familie innerhalb einer Generation eine Vollmutation entstand, hatte 59 Tripletts. Ein Fehlen der eingeschobenen AGG-Tripletts bei langen Prämutationen wird für die Instabilität zur Vollmutation hin verantwortlich gemacht. In Einzelfällen sind auch de novo Punktmutationen des Gens bekannt, die die Funktionsweise des FMR1-Proteins beeinträchtigen.
Die Anzahl der Triplett-Wiederholungen steigt im Verlauf aufeinander folgender Meiosen. Dies erklärt auch die Zunahme der Schwere der Krankheit im Verlauf der Generationen (auch Antizipation genannt). Die molekulare Ursache für die Triplett-Expansion ist möglicherweise das Verrutschen (slippage) der neu synthetisierten DNA-Stränge an den Replikationsgabeln während der Meiose.
Vererbung
Das Fragiles-X-Syndrom ist ein erblich bedingtes Syndrom, welches entsprechend in einigen Familien gehäuft auftreten kann. Da die Genmutation, die dieser Besonderheit zugrunde liegt, nur am X-Chromosom auftritt, müsste die Vererbung eigentlich auch der anderer X-chromosomaler Erbgänge folgen, im Fall des FXS gibt es allerdings einige bislang nicht geklärte Abweichungen hiervon.
Der X-chromosomale Erbgang
Der X-chromosomale Erbgang beruht darauf, dass Frauen jeweils zwei X-Chromosomen, Männer jedoch immer nur eines besitzen. Entsprechend geben Frauen immer ein X-Chromosom an ihre Nachkommen weiter, Männer können entweder ein X-Chromosom oder ein Y-Chromosom vererben und entsprechend festlegen, ob die Nachkommen männlich oder weiblich sind. Im Falle von X-chromosomalen Mutationen ergibt sich dabei ein typischer Erbgang, der sich durch folgende Eigenschaften auszeichnet:
- Die Mutter zeigt in der Regel keine Symptome der Mutation, wenn die Veränderung des Gens nur auf einem ihrer beiden X-Chromosomen existiert und der Effekt durch das andere kompensiert wird.
- Die Mutter kann das fehlerhafte Gen mit einer Wahrscheinlichkeit von 50 % an ihre Nachkommen weitergeben. Dadurch können 50 % ihrer weiblichen Nachkommen wieder Trägerinnen werden, 50 % nicht. Wird das fehlerhafte Gen an einen männlichen Nachkommen weitergegeben, so prägt es sich bei diesem aufgrund der fehlenden Kompensation aus.
- Der Vater kann das fehlerhafte Gen niemals an seine männlichen Nachkommen vererben, da diese von ihm das Y-Chromosom erhalten.
Aus diesen Gründen treten die Symptome einer X-chromosomalen Mutation wie etwa der Bluterkrankheit vor allem bei Männern auf (Hemizygotie), während Frauen häufiger nur Überträgerinnen des fehlerhaften Gens sind. Nur im ungünstigen Fall, dass die Töchter sowohl von der Mutter als auch vom Vater ein mutiertes Gen erhalten, prägt sich die Mutation auch bei den Frauen aus.
Abweichungen vom X-chromosomalen Erbgang
Beim Fragiles-X-Syndrom gibt es jedoch einige Besonderheiten, die nicht dem oben beschriebenen Erbgang entsprechen:
- Nicht alle Männer, an die das fehlerhafte Gen übertragen wurde, bekommen das Fragiles-X-Syndrom, etwa 20 Prozent bleiben symptomfrei.
- Etwa 30 Prozent der Frauen, die Trägerinnen sind, bekommen das Syndrom, obwohl sie eine unmutierte Genversion besitzen. Bei ihnen prägen sich die Symptome jedoch meist weniger stark aus.
- Das Syndrom kann in Familien phasenweise verstärkt, generationenlang jedoch auch gar nicht auftreten, obwohl es vererbt wird.
Die Gründe für diese Besonderheiten im Erbgang sind bislang ungeklärt. Gemeinhin wird versucht, sie durch äußere Einflüsse auf das Gen zu erklären.
Beratungsgrundlagen
Für die genetische Beratung ergeben sich somit folgende Konstellationen:
- Männliche Überträger mit einer FMR1-Prämutation sind nicht beeinträchtigt. Dasselbe gilt für ihre Töchter, die in jedem Fall das prämutierte Allel (in der Regel unverändert) erben werden. Da Söhne das Y-Chromosom erhalten, sind sie nicht betroffen.
- Männliche Träger einer Vollmutation sind betroffen; das klinische Bild kann von leichten bis schweren Erkrankungen reichen. Sie sind fertil, wenn sie auch selten Kinder zeugen. In ihren Spermien befindet sich entgegen der zu erwartenden Vollmutation nur ein Allel mit Prämutation, welches sie auf ihre Töchter übertragen.
- Weibliche Überträgerinnen einer FMR1-Prämutation sind nicht beeinträchtigt. Ihre Söhne oder Töchter können am Fragiles-X-Syndrom erkranken. Die Wahrscheinlichkeit hierfür ist von der Anzahl der CGG-Wiederholungen abhängig. In einer humangenetischen Beratung kann das Risiko anhand von statistischen Tabellen abgeschätzt werden. Diese Zahlen sind jedoch mit Vorsicht zu beurteilen, da sie nur an kleinen Patientenkollektiven gewonnen wurden.
- Weibliche Träger einer Vollmutation sind zum Teil nicht betroffen. Bei der Mehrzahl liegen jedoch geistige Beeinträchtigungen vor, deren Schwere im Vergleich zu den Männern häufig geringer ausgeprägt ist. Dies wird durch die zufällige Inaktivierung eines der beiden X-Chromosomen erklärt. Sie können entweder das gesunde oder das krankhafte X-Chromosom an ihre Nachkommen weitergeben. Die Wahrscheinlichkeit, dass diese Vollmutation an die Kinder weitergegeben wird, liegt somit bei 50 %. Die Kinder können in ihrem klinischen Bild erheblich von der Mutter abweichen.
Neurobiologie des Fragiles-X-Syndroms
Zur Erforschung der neurobiologischen Grundlagen, die die Symptomatik des Fragiles-X-Syndroms hervorrufen, wurde ein Tiermodell entwickelt. Dabei handelt es sich um eine sogenannte FMR1-Knockout-Maus, einen Mausstamm, bei dem durch geeignete molekularbiologische Methoden gezielt das FMR1-Gen entfernt wurde. Die Deletion von FMR1 in Mäusen ist von einigen Symptomen begleitet, wie sie auch für FXS-Patienten charakteristisch sind. Dazu gehören die Hyperaktivität, die epileptischen Anfälle und die Vergrößerung der Hoden. Im Gegensatz dazu waren Beobachtungen eines geringeren Lernvermögens der FMR1-Knockout-Mäuse nicht direkt auf den Menschen übertragbar, auch wegen der geringen Vergleichbarkeit der kognitiven Fähigkeiten zwischen Mäusen und Menschen.
Neuere Befunde zeigen jedoch, dass eine einfache Form des assoziativen Lernens, und zwar der klassischen Konditionierung, die beim Menschen und bei Mäusen gleichermaßen anzutreffen ist und den gleichen Mechanismen gehorcht, sowohl in FMR1-Knockout-Mäusen als auch FXS-Patienten schwer beeinträchtigt ist. Dabei handelt es sich um die Konditionierung des Lidschlussreflexes.
Beim Lidschlussreflex handelt es sich um einen Schutzreflex des Augenlides. Er wird aktiviert, wenn ein unangenehmer oder schmerzhafter Reiz auf die Oberfläche des Augapfels trifft. Das Augenlid schließt sich. Die Konditionierung des Lidschlussreflexes im Experiment geschieht folgendermaßen: Als sogenannter unkonditionierter Reiz dient ein kurzer Luftstoß auf den Augapfel. Der konditionierte Reiz ist ein Ton, der vor dem Luftstoß beginnt und gemeinsam mit ihm endet (beim sogenannten delay conditioning). Nach einigen Versuchen mit gleichem Intervall zwischen Beginn des Tones und dem Luftstoß schließt sich das Auge exakt zu einem Zeitpunkt, der gewährleistet, dass es beim Auftreffen des Luftstoßes bereits geschlossen ist. Konditioniert wird dabei das Timing des Reflexes.Die Konditionierung des Lidschlussreflexes kann sowohl im Tierversuch als auch am Menschen durchgeführt werden. Die neuronalen Schaltkreise, die für eine korrekte Anpassung des Reflexes verantwortlich sind, sind sehr gut bekannt und eingehend untersucht. Die beteiligten Neurone befinden sich im Kleinhirn. Von zentraler Bedeutung für die Konditionierung des Lidschlussreflexes ist eine Form der synaptischen Plastizität an der Parallelfasersynapse der Purkinjezellen. Der Befund, dass die Konditionierung des Lidschlussreflexes in FXS-Patienten verschlechtert ist, kann durchaus therapeutische Bedeutung gewinnen. Man könnte den Lidschlussreflex als Parameter verwenden, um die Wirksamkeit möglicher Therapien ganz objektiv zu messen.
Corticale Nervenzellen der FMR1-Knockout-Mäuse sowie der FXS-Patienten weisen eine erhöhte Anzahl und größere durchschnittliche Länge der Dornfortsätze (sog. spines) auf. Das lässt auf eine synaptische Funktion des FMR1-Proteins schließen. Das FMR1-Protein ist ein RNA-bindendes Protein. Man nimmt heute an, dass eine seiner Funktionen darin besteht, die Translation der gebundenen RNA solange zu hemmen, wie diese unterwegs vom Zellkern im Perikaryon zum Dendriten ist. Dort funktioniert das FMR1-Protein dann als eine Art Schalter, der die RNA freigibt und deren Translation als Antwort auf synaptische Signale ermöglicht. Demnach gehört das FMR1-Protein zu den Faktoren, die für eine aktivitätsabhängige Proteinsynthese an Synapsen erforderlich sind.
Die mGluR-Theorie des FXS
Das FMR1-Protein wird an Synapsen nach Aktivierung metabotroper Glutamatrezeptoren (mGluR) synthetisiert. Die Gruppe-1-mGluR stimulieren einerseits die Proteinsynthese, andererseits aber auch den Transport FMR1-Protein-assoziierter RNA in den Dendriten. Das legt die Vermutung nahe, dass das FMR1-Protein hemmend auf die Synthese anderer synaptischer Proteine wirkt. Damit übereinstimmend wurde gefunden, dass im Hippocampus bestimmte Formen der synaptischen Plastizität, die abhängig von Proteinsynthese sind, in den FMR1-Knockout-Mäusen verstärkt sind, während andere Formen, die unabhängig von der Synthese von Proteinen sind, unverändert bleiben. Daraus wurde geschlussfolgert, dass auch andere Formen der mGluR- und proteinsyntheseabhängigen synaptischen Plastizität durch Deletion des FMR1-Gens hochreguliert sein müssten. In der Tat war das auch in Purkinjezellen der Fall. Dort ist die Langzeitdepression an der Parallelfasersynapse ebenfalls mGluR-abhängig und erwies sich als verstärkt in den FMR1-Knockout-Mäusen. Die spines der Purkinjezellen sind in FMR1-Knockout-Mäusen verlängert. Alle diese Veränderungen legen nahe, dass das FMR1-Protein als Regulator der synaptischen Struktur sowie der mGluR-abhängigen Plastizität wirkt.
Behandlung
Aufgrund der genetischen Ursache ist eine Heilung nicht möglich. Symptomatisch kann nach eingehender kinderpsychiatrischer, pädiatrischer und neurologischer Untersuchung ein individuelles Förderprogramm erstellt werden, welches Verhaltenstherapie, Ergotherapie, Musiktherapie und logopädische Betreuung einschließt. Diese Programme können sehr erfolgreich sein, wenn ein günstiges Umfeld hergestellt wird. Weiterhin können in Deutschland über die zuständigen Gesundheitsämter sowie die Kreissozialämter als Kostenträger „Maßnahmen zur Wiedereingliederung“ nach SGB XII §§ 53 ff beantragt werden.
Literatur
Wissenschaftliche Literatur
- J. P. Martin, J. Bell: A pedigree of mental defect showing sex-linkage. in: Journal of neurology, neurosurgery, and psychiatry (J. Neurol. Psychiat.). BMJ Publishing Group, London 6.1943, 154-157. ISSN 0022-3050
- H. A. Lubs Jr.: A marker X chromosome. in: American journal of human genetics (Am. J. Hum. Genet.). Univ. of Chicago Press, Chicago Ill. 21.1969, 231-244. ISSN 0002-9297
- B. Beek, P. B. Jacky, G. R. Sutherland: Heritable fragile sites and micronucleus formation. in: Annales de Genetique. 26.1983, 5-9.
- P. B. Jacky, B. Beek, G. R. Sutherland: Fragile sites in chromosomes: Possible model for the study of spontaneous chromosome breakage. in: Science. 220.1983, 69-70.
- E. J. Kremer, M. Pritchard, M. Lynch, S. Yu, K. Holman, E. Baker, S. T. Warren, D. Schlessinger, G. R. Sutherland, R. I. Richards: Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. in: Science. 252.1991, 1711-1714. ISSN 0036-8075 (Abstract)
- B. A. Oostra, P. Chiurazzi: The fragile X gene and its function. in: Clinical genetics. An international journal of genetics in medicine (Clin. Genet.). Blackwell Munksgaard, Oxford 60.2001, 399-408. ISSN 0009-9163 (Abstract)
- A. L. Reiss, E. Aylward, L. S. Freund, P. K. Joshi, R. N. Bryan: Neuroanatomy of fragile X syndrome, the posterior fossa. in: Annals of neurology (Ann Neurol). Wiley-Liss, New York NY 29.1991,1(Jan), 26-32. ISSN 0364-5134 (Abstract)
- A. J. Verkerk, M. Pieretti, J. S. Sutcliffe, Y. H. Fu, D. P. Kuhl, A. Pizzuti, O. Reiner, S. Richards, M. F. Victoria, F. P. Zhang: Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. in: Cell. Massachusetts Institute of Technology. Cell Press, Cambridge Mass 31;65.1991,5 (May), 905-14. ISSN 0092-8674 (Abstract)
Weitere Literatur
- Ursula G. Froster (Hrsg.): Das Fragile-X-Syndrom. Quintessenz-Verlag, Berlin 1998, ISBN 3-8208-1764-6
- Suzanne Saunders: Das Fragile X-Syndrom - Ein Ratgeber für Fachleute und Eltern. Verlag der Bundesvereinigung Lebenshilfe für Menschen mit geistiger Behinderung e.V., Marburg 2003. ISBN 3-88617-306-2
Siehe auch
Weblinks
- Fragiles-X-Syndrom bei Online Mendelian Inheritance in Man
- The National Fragile X Foundation (Englisch)
- Interessengemeinschaft Fragiles-X e.V. (eine deutsche Selbsthilfegruppe)
Einzelnachweise

Bitte den Hinweis zu Gesundheitsthemen beachten! 

Dieser Artikel wurde am 5. Juli 2004 in dieser Version in die Liste der exzellenten Artikel aufgenommen.

Wikimedia Foundation.