- Morbus Alexander
-
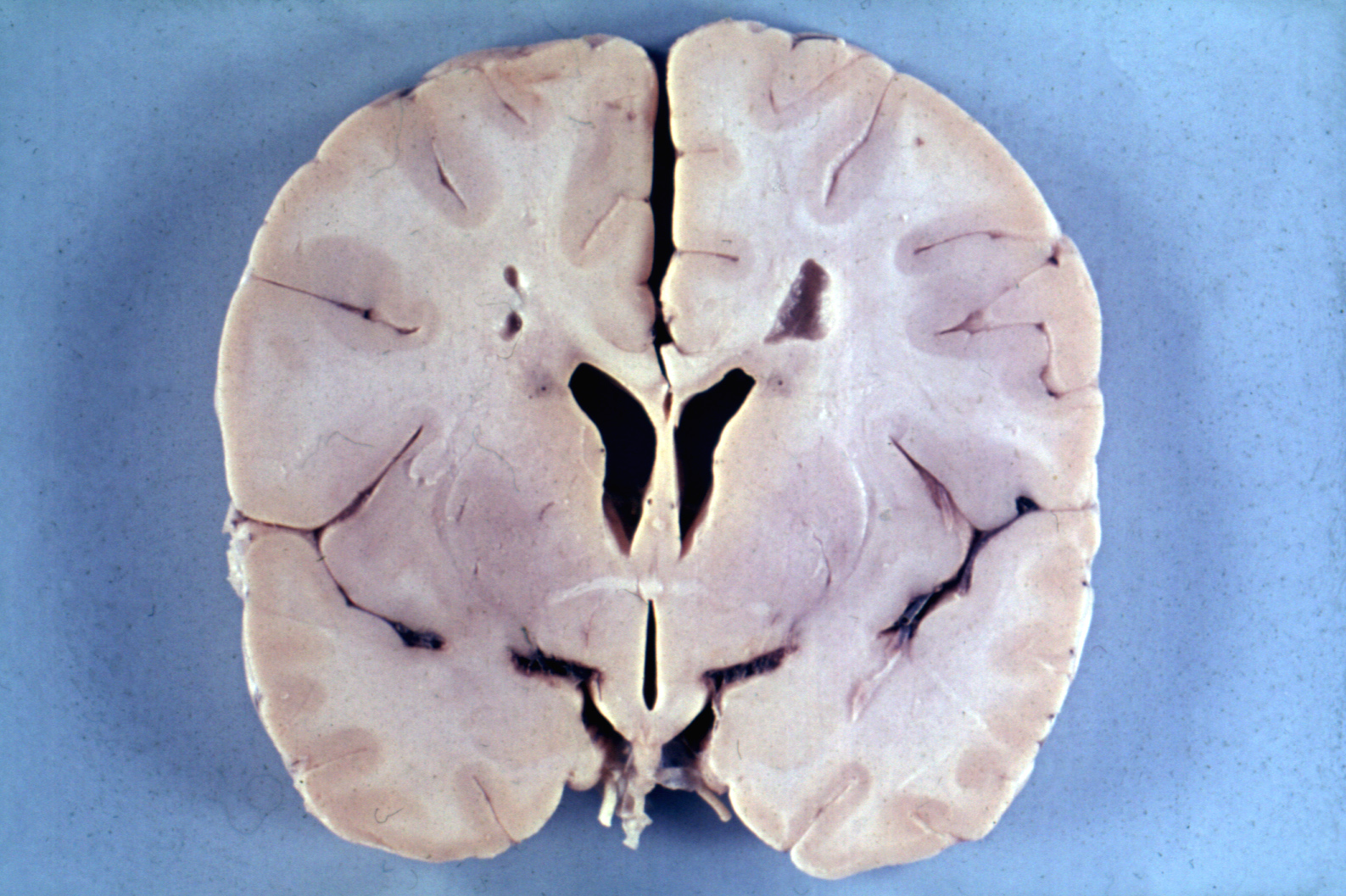
Klassifikation nach ICD-10 G37.8 Sonstige näher bezeichnete demyelinisierende Krankheiten des Zentralnervensystems ICD-10 online (WHO-Version 2006)  Gehirn bei Alexander-Krankheit.
Gehirn bei Alexander-Krankheit.
Autopsiefall eines vierjährigen Jungen mit übermäßiger Größe des Gehirns (Makroenzephalie) und periventrikulärer Entmarkung.Die Alexander-Krankheit (Morbus Alexander) ist eine seltene Erkrankung aus der Gruppe der Leukodystrophien. Es handelt es sich um eine genetisch bedingte Störung, bei der eine fortschreitende Degeneration der weißen Substanz von Gehirn und Rückenmark auftritt. Klinisch macht sich die Alexander-Krankheit häufig bereits im Kleinkindesalter mit einer Verzögerung der psychomotorischen Entwicklung und einer Zunahme der Schädelgröße bemerkbar. Ursache der bei der Erkrankung auftretenden strukturellen Veränderungen der Stützzellen des Gehirns (Astrozyten) und einer Entmarkung sind meist spontan auftretende dominante Mutationen des GFAP-Gens, das für ein astrozytäres Strukturprotein, das saure Gliafaserprotein, kodiert. Eine Heilung der Krankheit ist nicht möglich. Erkrankte Kleinkinder überleben in der Regel das sechste Lebensjahr nicht.
Inhaltsverzeichnis
Geschichtliche Aspekte
Benannt wurde die Erkrankung nach dem neuseeländischen Pathologen William Stewart Alexander, der als junger Arzt in London bei der Neuropathologin Dorothy Stuart Russell (1895–1983) den Sektionsfall eines 15 Monate alten Kindes aufgearbeitet hatte[1] und das Krankheitsbild 1949 erstmals beschrieb.[2]
Epidemiologie
Die Mehrzahl der Krankheitsfälle tritt sporadisch auf, also ohne familiäre Häufung. Bei der Mehrzahl der Patienten beginnt die Alexander-Krankheit im Kleinkindesalter, wobei Mädchen etwas häufiger als Jungen zu erkranken scheinen. Die Krankheit ist sehr selten und konnte bislang bei etwa 150 Patienten molekulargenetisch bestätigt werden.[3]
Klinisches Bild
Abhängig vom Manifestationsalter lassen sich verschiedene Formen der Alexander-Krankheit abgrenzen.
Die bei Kindern auftretende infantile Form ist am häufigsten. Die Erkrankung macht sich im Alter von ungefähr sechs Monaten bemerkbar, kann aber auch zwischen dem ersten und 24. Lebensmonat einsetzen. Unspezifisches Leitsymptom bei betroffenen Kindern ist eine ausgeprägte Störung der motorischen und geistigen Entwicklung. Eine Vergrößerung des Schädels (Makrozephalus) und des Gehirns (Makroenzephalie), Probleme beim Füttern, Schluckstörungen, Ataxie, Spastizität und Krampfanfälle können hinzutreten.
Die neonatale Form (bei Neugeborenen) zeigt den am raschesten fortschreitenden Verlauf mit der schlechtesten Prognose.
Die juvenile Form (bei Jugendlichen) verläuft milder, die Patienten haben nicht immer eine Makrozephalie und die neurologischen Defizite treten später auf.
Bei der adulten Form, die bei Erwachsenen auftritt, ist das Ausmaß degenerativer Veränderungen der weißen Substanz geringer und es steht häufig eine bulbäre Symptomatik mit Schluck- und Sprechstörungen im Vordergrund, da der Hirnstamm besonders betroffen ist. Myoklonien des Gaumensegels können auftreten.[4]
Diagnostik
Die Erkrankung kann nicht klinisch diagnostiziert werden, das heißt nicht alleine aufgrund einer körperlichen Untersuchung. Vielmehr sind technische Untersuchungsverfahren notwendig. Bis vor wenigen Jahren war ein bioptischer oder autoptischer Nachweis der für die Alexander-Krankheit charakteristischen pathologischen Veränderungen im Hirngewebe zur Diagnosesicherung notwendig. Seit der Charakterisierung des verantwortlichen Gens werden in Verdachtsfällen zunehmend DNA-Analysen durchgeführt. Ebenso kann eine kernspintomographische Untersuchung des Gehirns die Abgrenzung gegenüber anderen Erkrankungen erlauben. Bei den kindlichen Verlaufsformen wurden für die Diagnosestellung folgende neuroradiologische Kriterien vorgeschlagen:
- ausgedehnte frontale Marklagerveränderungen.
- ein periventrikulärer Saum (in der T1-Wichtung signalintensiv).
- ein periventrikulärer Saum (in der T2-Wichtung signalarm).
- Veränderungen in Basalganglien, Thalamus und Hirnstamm.
- Kontrastmittelanreicherung in bestimmten Hirnregionen.
In Anwesenheit von vier der fünf Kriterien gilt die die Diagnose einer Alexander-Krankheit als wahrscheinlich.[5] Bei der adulten Form lassen sich eine Atrophie und Signalveränderungen im Bereich von Hirnstamm und Rückenmark nachweisen.[6]
Pathologie
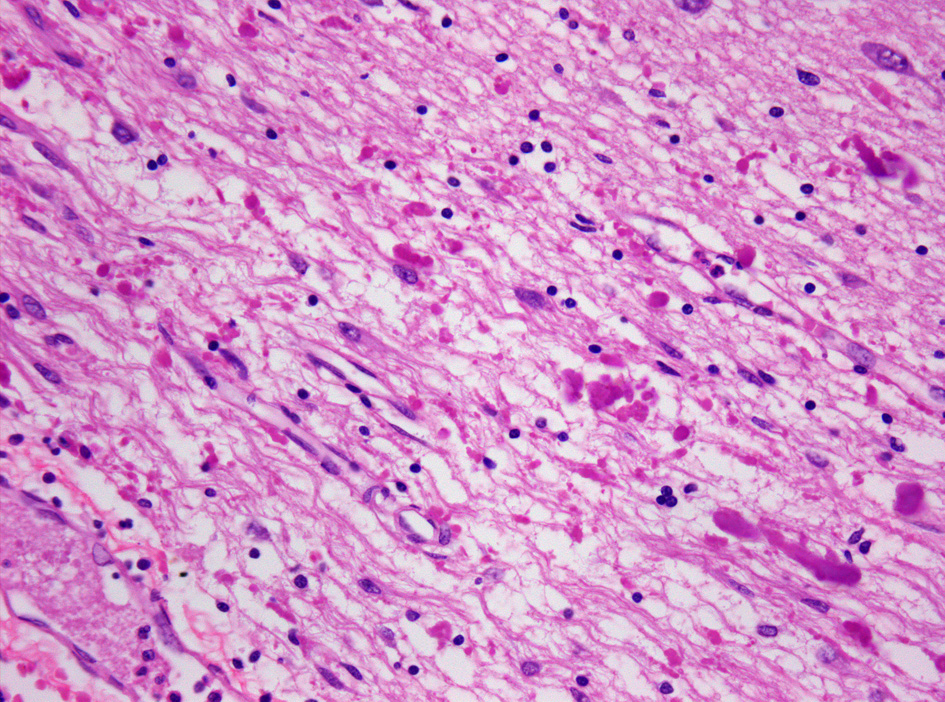
 Histopathologie der Alexander-Krankheit mit Nachweis zahlreicher plumper Rosenthal-Fasern im periventrikulären Marklager.
Histopathologie der Alexander-Krankheit mit Nachweis zahlreicher plumper Rosenthal-Fasern im periventrikulären Marklager.
Hämatoxylin-Eosin-gefärbtes Autopsiegewebe. Vergrößerung 400-fachDie Alexander-Krankheit ist primär eine Erkrankung der Astrozyten, Stützzellen des Gehirns (Glia), deren Intermediärfilament GFAP fehlerhaft gebildet wird. Zusammengelagert mit anderen Proteinen bilden sich astrozytäre Einschlüsse, die als Rosenthal-Fasern bekannt sind. Feingeweblich sind Rosenthal-Fasern bei der Erkrankung im gesamten Zentralnervensystem (Gehirn und Rückenmark) nachweisbar, finden sich jedoch bevorzugt periventrikulär und in der Umgebung von Blutgefäßen. Elektronenmikroskopisch zeigt sich eine enge Verbindung der Rosenthal-Fasern mit Intermediärfilamenten.
Darüber hinaus findet sich bei Kindern eine mangelhafte Myelinisierung und bei älteren Patienten eine Entmarkung. Dabei sind sowohl sensorische als auch motorischen Nervenfasern betroffen. Die Myelinisierungsstörung wird insbesondere bei kleinen Kindern häufig von einer Makrozephalie und manchmal von einem Hydrozephalus begleitet.
Das Verteilungsmuster der entmyelinisierten Areale stimmt nicht mit dem der Rosenthal-Fasern überein; Myelinisierungsstörung und Faserbildung scheinen also voneinander unabhängige Manifestationen der Krankheit zu sein. Es wird angenommen, dass Veränderungen der Astrozyten sekundär zu einer Myelinisierungsstörung führen.
Genetik
Die autosomal dominante Alexander-Krankheit wird meist durch spontan auftretende Mutationen des GFAP-Gens verursacht, das für ein astrozytäres Intermediärfilament, das saure Gliafaserprotein kodiert.[7] In etwa 94 % der Fälle lässt sich eine GFAP-Mutation nachweisen, wobei eine Vielzahl verschiedener Mutationen beschrieben worden sind. Diese betreffen meist Veränderungen von Aminosäure-Resten, die evolutionär hoch konserviert und für die Dimerisation der Proteine und Formation von Mikrofilamenten verantwortlich sind.[8][9] Offenbar besteht ein Zusammenhang zwischen Art der genetischen Veränderung und Ausprägung der Erkrankung, dem Phänotyp: Bei Patienten mit einer das Codon 79 betreffenden Punktmutation (R79H) sind im allgemeinen weniger schwer erkrankt, als Patienten bei denen das Codon 239 betroffen ist.[9]
Es sind einzelne Familien beschrieben worden, in denen mehrere Kinder gesunder Eltern erkrankten.[10] In diesen seltenen Fällen liegt vermutlich entweder ein autosomal-rezessiver Erbgang vor oder ein Keimbahnmosaizismus mit Weitergabe der dominanten Mutation in Zellen der Keimbahn der gesunden Eltern. Tatsächlich scheint die GFAP-Mutation überwiegend das vom Vater stammende Allel zu betreffen, was eine Entstehung der Mutation während der Spermienbildung nahelegt.[11] Eltern betroffener Kinder ist darum bei fortbestehendem Kinderwunsch eine genetische Beratung empfohlen worden.[9]
Prognose und Therapie
Trotz der Aufklärung der zugrunde liegenden Mutation ist bislang keine Heilung möglich und die Behandlung der Alexander-Krankheit daher ausschließlich symptombezogen und unterstützend. Kinder, die die Alexander-Krankheit als Kleinkinder entwickeln, überleben in der Regel das sechste Lebensjahr nicht.
Literatur
- Rodriguez: Alexander disease. Orphanet Encyclopedia. 2004. (Übersichtsarbeit) Volltext als PDF
- Harding & Surtees: Metabolic and neurodegenerative disease of childhood. In: Greenfield´s Neuropathology. 7. Auflage. 2002. Arnold, London ISBN 0-34074231-3
Einzelnachweise
- ↑ Scolding. Brain, 2004;127:2144-2147
- ↑ Alexander: Progressive fibrinoid degeneration of fibrillary astrozytes associated with mental retardation in a hydrozephalic infant. Brain 1949; 72:373-381; PMID 15409268 (Erstbeschreibung).
- ↑ Zusammenstellung des Waisman-Center anhand dieser Bibliographie.
- ↑ Pareyson et al.: Adult-onset Alexander disease: a series of eleven unrelated cases with review of the literature. Brain. 2008;131(Pt 9):2321-2331; PMID 18684770.
- ↑ van der Knaap et al.: Alexander disease: diagnosis with MR imaging. AJNR Am J Neuroradiol. 2001;22(3):541-552; PMID 11237983; Volltext.
- ↑ Farina et al.: Can MR imaging diagnose adult-onset Alexander disease? AJNR Am J Neuroradiol. 2008;29(6):1190-1196; PMID 18388212.
- ↑ Brenner et. al.: Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander Disease. Nature Genetics 2001; 27:117–120; PMID 11138011.
- ↑ Gorospe: Alexander disease. Gene Reviews; Volltext.
- ↑ a b c Rodriguez et al.:Infantile Alexander disease: spectrum of GFAP mutations and genotype-phenotype correlation. Am J Hum Genet. 2001;69(5):1134-1140; PMID 11567214; Volltext.
- ↑ Wohlwill et al.: Dysmyelinogenic leukodystrophy; report of a case of a new, presumably familial type of leukodystrophy with megalobarencephaly. J Neuropathol Exp Neurol. 1959;18(3):359-383; PMID 13665382.
- ↑ Li et al.: Propensity for paternal inheritance of de novo mutations in Alexander disease. Hum Genet. 2006;119:137-144; PMID 16365765.
Weblinks
- Alexander-Krankheit bei Online Mendelian Inheritance in Man
- Alexander-Krankheit in Orphanet
- Ressourcen zu Erforschung und Molekulargenetik der Alexander-Krankheit der University of Wisconsin-Madison
Wikimedia Foundation.
Schlagen Sie auch in anderen Wörterbüchern nach:
Morbus — ist das lateinische Wort für Krankheit. In der Sprache der Medizin gibt Morbus in Verbindung mit dem Namen des Erstbeschreibers einer Erkrankung einen Namen (siehe: Eponym), wobei oft verschiedene Namen für die gleiche Erkrankung üblich sind. Im… … Deutsch Wikipedia
Alexander-Krankheit — Klassifikation nach ICD 10 E75.2 Sonstige Sphingolipidosen … Deutsch Wikipedia
Alexander Strauch — (* 22. Dezember 1971 in München) ist ein deutscher Komponist. Inhaltsverzeichnis 1 Ausbildung 2 Werk 3 Werkauswahl 4 Weblinks … Deutsch Wikipedia
Alexander Dolgopolow — Spitzname: Dolgo, The Do … Deutsch Wikipedia
Alexander Gordon Bearn — (* 29. März 1923 in Cheam, Surrey, England; † 15. Mai 2009 in Philadelphia, Pennsylvania, Vereinigte Staaten) war ein britisch amerikanischer Mediziner und Genetiker. Er beschäftigte sich mit der Erforschung des Morbus Wilson, wofür er neue… … Deutsch Wikipedia
Alexander Bittorf — (voller Name Eduard August Alexander Bittorf; * 29. April 1876 in Leipzig; † 20. Februar 1949 ebenda) war ein deutscher Internist und Pathologe. Er lehrte an den Universitäten Breslau und Leipzig. Inhaltsverzeichnis 1 Leben 2 Wirken … Deutsch Wikipedia
Alexander Solomon Wiener — (* 16. März 1907 in Brooklyn, New York, USA; † 7. November 1976 in New York) war ein US amerikanischer Serologe. Er entdeckte 1937 zusammen mit Karl Landsteiner den Rhesusfaktor. 1946 erhielt er den Albert Lasker Award for Clinical Medical… … Deutsch Wikipedia
Alexander von Winiwarter — (* 22. April 1848 in Wien; † 31. Oktober 1917 in Lüttich) war ein österreichisch belgischer Chirurg. Inhaltsverzeichnis … Deutsch Wikipedia
Morbus Wilson — Klassifikation nach ICD 10 E83.0 Morbus Wilson … Deutsch Wikipedia
Morbus Menière — Klassifikation nach ICD 10 H81.0 Menière Krankheit … Deutsch Wikipedia
 18+© Academic, 2000-2026
18+© Academic, 2000-2026- Kontaktieren Sie uns: Unterstützung, Werbung
Wörterbücher Export, schritte mit PHP, Joomla, Drupal, WordPress, MODx.