- Myasthenia gravis pseudoparalytica
-
Klassifikation nach ICD-10 G70 Myasthenia gravis und sonstige neuromuskuläre Krankheiten G70.0 Myasthenia gravis ICD-10 online (WHO-Version 2006) Die Myasthenia gravis pseudoparalytica (von griech. mys „Muskel“, -asthenie „Schwäche“, lat. gravis „schwer“ pseudo „falsch“ und paralysis „Lähmung“; Kürzel: MG) ist eine seltene neurologische Erkrankung, deren charakteristisches Kennzeichen eine abnorme belastungsabhängige Muskelschwäche ist, die sich in Ruhe wieder bessert. Sie macht sich durch eine schnelle Ermüdbarkeit bei wiederholten Bewegungen bemerkbar.
Die kausale Basis der Erkrankung ist die gestörte (und im Verlauf zerstörte) Signalübertragung zwischen Nerv und Muskel. Es handelt sich wahrscheinlich um eine Autoimmunerkrankung der motorischen Endplatte der quergestreiften Muskulatur. Polyklonale IgG-Antikörper binden an den nikotinischen Acetylcholinrezeptor und blockieren die Weiterleitung des Aktionspotenzials auf die Muskelzelle.
Inhaltsverzeichnis
Myasthenia gravis beim Menschen
Beschwerdebild (Symptomatik)
 Der sog. „Schlafzimmerblick“ bei Myasthenia gravis
Der sog. „Schlafzimmerblick“ bei Myasthenia gravisAm Morgen und nach Ruhepausen ist die Leistungsfähigkeit am besten, doch nach wenigen wiederholten Bewegungen sind verschiedene Muskeln oder auch ganze Muskelgruppen erschöpft. Häufig treten die Symptome abends stärker auf. Von den Lähmungen bzw. anfänglich nur Einschränkungen der Beweglichkeit sind besonders kleine Muskeln betroffen, prinzipiell können aber alle quergestreiften (willkürlich bewegbaren) Muskeln betroffen sein.
Muskelgewebe ohne motorische Endplatten, wie der Herzmuskel und die glatte Muskulatur, sind nicht von der Krankheit betroffen. Die Beteiligung Letzterer ist allerdings wissenschaftlich bisher nie ausgeschlossen worden. Ein Hinweis auf Beteiligung könnte die Wirksamkeit der Pyridostigmin-basierten Myasthenie-Medikamente auf die Darmtätigkeit sein, da der Darm (bis auf Schließmuskulatur) aus glatter Muskulatur besteht. Ein weiterer Hinweis könnte auch die darmverursachte Obstipation sein, die zu den Symptomen des ebenfalls autoimmun-verursacht vermuteten Lambert-Eaton-Rooke-Syndrom (LEMS) gehört.
Häufig macht sich die Myasthenia gravis zuerst an den Augen, Augenlidern und den äußeren Augenmuskeln bemerkbar: Durch Ermüden der Lidhebermuskeln kommt es zum typischen „Schlafzimmerblick“ (Ptosis), weil die Lider nicht mehr hochgehalten werden können. Die Patienten legen den Kopf zurück, um unter den Lidern hindurchschauen zu können. Typische Frühsymptome sind daher Ptosis sowie Diplopie (Doppelbilder), aber auch eine mangelnde oder unmögliche Schließbarkeit eines oder beider Augenlider. Weitere typischerweise früh betroffene Muskelgruppen sind die mimische Muskulatur, die Mund- und Zungenmuskulatur („lachendes“ starres Gesicht, verwaschenes oder unmögliches Sprechen), die Hals- und Nackenmuskulatur (Kopfhalteschwäche), die Kau- und die Rachenmuskulatur (Schluckstörung bis hin zur Unfähigkeit Speichel abzuschlucken). Bei weiterem Fortschreiten sind in den meisten Fällen die Arme stärker betroffen als die Beine. Ferner kann die Atemmuskulatur so stark beeinträchtigt werden, dass Betroffene nur im Sitzen schlafen können oder in fortgeschrittenem Stadium beatmet werden müssen. Auch die Schluckmuskulatur kann so stark betroffen sein, dass eine unterstützende oder vollständige Versorgung über eine Magensonde indiziert ist.
Gleichgewichts-, degenerative Gedächtnis- oder Sensibilitätsstörungen sind keine Anzeichen einer MG.
Zur Charakteristik der Symptome ist zu sagen, dass sie wechselnd, nicht statisch und starr sind. Wechselnd sowohl in Bezug auf Uhrzeit, auslösende Stärke der Belastung und der „Auswahl“ aktuell betroffener Muskeln bzw. Muskelgruppen – z. B. linke Hand rechtes Auge, einmal zusammen, einmal einzeln oder früh oder abends. Die Symptome sind nicht vorhersehbar und meist plötzlich „hereinbrechend“. Manche Muskeln bzw. Muskelgruppen sind lediglich in ihrer Beweglichkeit eingeschränkt, bei anderen kann wiederum die ganze Kraft fehlen, sie überhaupt zu bewegen. Da diese Wechselhaftigkeit des Verlaufs bei einem einzelnen Betroffenen bzw. im Vergleich von Patient zu Patient so vielfältig ist, kann eigentlich von einer generellen Klinik (Erscheinungsform) nicht gesprochen werden.
Erstsymptomatik
- Über 50 % beginnend mit okulärer Symptomatik (z. B. Lidschwäche und/oder Doppelbilder)
- Etwa 14 % beginnend mit Schluck- und Sprechstörungen
- Etwa 8 % beginnend mit Arm- bzw. Beinschwächen (z. B. unklarer Fußheber-Paresen)
- sehr seltener Beginn bei der Rumpf- und Wirbelsäulenmuskulatur
Komplikationen
Lebensbedrohlich ist in einer Krise das akute Versagen der Atemmuskeln. Bei dieser akuten Verschlechterung der Symptome mit Atemproblemen spricht man von einer „myasthenen Krise“ oder bei Überdosierung von Cholinesterasehemmern wie Pyridostigmin, von einer „cholinergenen Krise“. Sehr hilfreich ist hier der Nothilfe-Ausweis, der medizinische Maßnahmen und Ansprechpartner beschreibt. (Ausweise werden von der „Deutschen Myasthenie Gesellschaft“ bzw. von betreffenden Pharmaherstellern ausgegeben). Im Falle beider Krisen ist rasche Aufnahme und kompetente Behandlung auf einer Intensivstation gefordert, da die Mortalitätsrate immer noch recht hoch ist (nach Literatur 4–13 %).
Durch Verschlucken (Schluckstörung) kann es zu einer schweren Lungenentzündung kommen, einer Aspirationspneumonie.
Symptomverstärkend sind:
- Infekte, Entzündungen, Fieber, Hitze, Vibrationen
- Interkurrente Erkrankungen (Schilddrüse)
- extreme Belastungen (seelisch, körperlich)
- auch Belastungen, wie Narkosen, vor allem bei der Wahl myasthenie-kontra-indizierter Narkotika
- hormonelle Schwankungen
- Fehlbehandlungen bzw. -dosierungen bei der medikamentösen Therapie (das Zuviel oder Zuwenig, was symptomatisch auch schwankt)
- Verschiedene Medikamente (myasthenie-kontra-indizierte Schmerzmittel, Antibiotika, Psychopharmaka, Hormonprodukte usw.)
Eine nicht unerhebliche Komplikation entsteht ebenfalls mit der Tatsache, dass die Diagnose MG sehr spät (statistisch nach 2,7 Jahren) gestellt wird. Aufgrund erwähnter Wechselhaftigkeit der Symptome, sowie der anfänglichen schwachen Ausprägung von Symptomen, werden diese häufig übersehen oder fehlinterpretiert, so dass es sogar zu erheblichen diagnostischen Missverständnissen und Fehldiagnosen kommt. Einerseits bedeutet das für die Betroffenen zusätzliche Symptomverschlechterung aufgrund des Fehlens der dringlichen medikamentösen Behandlung sowie eine zusätzliche psychische Belastung wegen der nicht seltenen Odyssee durch medizinische Einrichtungen.
Historisches
MG ist seit dem 17. Jahrhundert bekannt. Der erste dokumentierte Fall wurde in englisch-amerikanischer Kolonialpost gefunden. Er stammt aus dem Jahr 1644 und beschreibt die Schwäche des Indianerhäuptlings Opechankanough zu gehen (Universität zu Virginia, Neurologie, Charlottesville). Englische Erstbeschreibung 1672 durch den Oxforder Arzt Thomas Wills im lateinisch verfassten „De Anima Brutorum“ (was allerdings bis 1903 unbeachtet blieb). Erste modernere Fallbeschreibung 1877, durch den Londoner Arzt Samual Wilks. Weitere deutsche Fallberichte im 19. Jahrhundert durch den Heidelberger Wilhelm Erb und Oppenheim. 1893 in Warszczawa (Warschau) durch Samuel Goldflam bisher umfassendste Publikation. Jolly führte nachweisende elektrophysiologische Untersuchungen durch und prägte ab 1895, den bis heute üblichen Namen „Myasthenia gravis pseudoparalytica“. Nach ersten neuen Erkenntnissen über den Zusammenhang mit der Thymusdrüse, Anfang des 20. Jahrhunderts führte 1912 Sauerbruch die erste Thymektomie durch. Ab 1934 gab es durch einen bisher unbekannten Konsiliarus und die englische Assistenzärtzin Mary Walker, die erste wirksame medikamentöse Therapie und wies damit auf eine gestörte Übertragung zwischen Nerv und Muskel hin. Dieser kausale Zusammenhang war allerdings erst nach 1960 nachgewiesen. Bereits 1960 wies John Simpson hypothetisch auf eine autoimmune Genese hin. Der Nachweis des AChR-Antikörper gelang erst 1974. [1]
Pathophysiologie
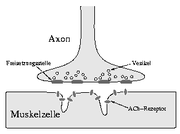 Muskelendplatte
MuskelendplatteDie Myasthenia gravis ist eine Autoimmunerkrankung, d. h. der Körper bildet Autoantikörper, welche mit den Acetylcholinrezeptoren (AChR) an der motorischen Endplatte eine reversible Bindung eingehen. Deshalb kann der elektrische Impuls (das Aktionspotenzial) vom Nerven nicht mehr auf den Muskel übertragen werden, der Muskel wird nicht erregt. Darüber hinaus verringert sich die Anzahl der Acetylcholinrezeptoren, da durch die Bindung der Antikörper an den Acetylcholinrezeptoren, die Acetylcholinrezeptoren durch eine Immunaktivität abgebaut werden. Dabei zerfällt die Struktur der subsynaptischen Membran in Bruchstücke. Durch Endozytose entsteht ein Autophagosom. Transportvesikel mit Verdauungsenzymen verschmelzen mit den Autophagosomen. Die Acetylcholinrezeptoren werden durch diese Immunreaktion alle zwei bis drei Tage abgebaut. Die Struktur der motorischen Endplatten wird verändert. Die subsynaptischen Einfaltungen (junctional folds) werden flacher und der synaptische Spalt wird breiter. Dies hat zur Folge, dass bei der Ausschüttung des Acetylcholins, Acetylcholin aus dem synaptischen Spalt diffundiert oder es von dem Enzym Cholinesterase hydrolysiert wird, bevor es einen Acetylcholinrezeptor besetzen kann. Die Erkrankung kann in jedem Alter auftreten, ist aber bei Frauen zwischen 20 und 40 Jahren gehäuft. Bei MG sind Veränderungen des Thymus sehr häufig nachweisbar. Man vermutet, dass der Thymus bei der Erkrankung eine wichtige Rolle spielt. Gelegentlich ist die MG durch ein Thymom bedingt, der die schädlichen Autoantikörper produziert. In diesen Fällen kann durch eine chirurgische Entfernung der Thymusdrüse die Krankheit geheilt werden.
Ursachen
Weder Ursachen noch Auslöser dieser Erkrankung sind heute vollständig erforscht bzw. zu erklären. Es wird vermutet, dass es neben den bekannten Antikörpern AChr und MuSK noch weitere Myasthenie-Relevante Antikörper geben könnte. Nachgewiesen sind lediglich bestehende Zusammenhänge zu Myasthenie-relevanten Antikörpern und zur Thymusdrüse und der kausalen Basis der Erkrankung, der gestörten Signalübertragung zwischen Nerv und Muskel. Ungeklärt ist auch der Auslöser der schwankenden Symptomatik bei Umwelteinflüssen, Infekten, Entzündungen, seelischen und psychischen Belastungen. [2]
Diagnose
Die Diagnose wird vor allem aus:
- klinischem Bild,
- dem Nachweis von Acetylcholinrezeptor-Antikörpern (AChR-AK),
- der sofortigen, kurzen (nur einige Minuten anhaltenden) Besserung nach Verabreichung von Acetylcholinesterase-Inhibitoren und durch
- elektrophysiologische Untersuchungen
gestellt. Abhängig von der Schwere der Erkrankungen unterteilt man die MG in verschiedene Stadien (nach Osserman und Genkins):
Stadium Bezeichnung befallene Körperregionen I rein okuläre Myasthenia gravis nur Augenmuskeln IIa leichte Generalisierung ganzer Körper IIb mittelschwere Generalisierung ganzer Körper III schwere akute Generalisierung ganzer Körper mit Atemmuskulatur IV schwere chronische Generalisierung Der „Besinger-Score“ dient der klinischen Verlaufsbeurteilung.
Einzelne Untersuchungsmethoden sind:
- Simpson-Test (Ermüden der Augenlider im längeren Aufblick)
- Suche nach Doppelbildern
- Intensive, über das „normale Maß“ der Standard-Neurologie hinausgehende Belastungstests verschiedener Muskelgruppen - Voraussetzung zur Erstellung eines aussagekräftigen MG-Score
- Prostigmintest, Tensilon-Test (vorübergehendes Verschwinden von Symptomen nach intravenöser Injektion), richtungsweisend für eine Neuro-Transmitter-Erkrankung, aber nicht alleinig, spezifisch für eine MG
- Repititive Serienstimulation einzelner Nerven (RSN) – elektropysiologischer Test, bei dem bei Betroffenen unter der künstlichen Simulation (Nachahmung) von Muskelbelastung, ein Dekrement² nachgewiesenen werden kann.
- Einzelfaser-EMG - ähnlich, wie die Stimulation elektropysiologisch, allerdings sehr aufwendig und im Falle der Seronegativen MG kein Nachweis möglich
- Elektromyographie (Dekrement zeichen - Abnahme der Amplitude der evozierten Reizantwortpotentiale im Erfolgsmuskel)
- Histologischer Nachweis von Lymphoidzellen in Muskelbioptaten
- Labor-Diagnostik - Nachweis von Acetylcholinrezeptor-Antikörpern und /oder von Anti-MuSK-Antikörpern im Blut
- Belastungstests im Rahmen einer Schluck-Diagnostik durch myasthenie-erfahrene Logopädische Therapeuten, apparativ mittels einer Videoendoskopie und/oder bildgebender Schluckstudie
- bildgebende Diagnostik (CT mit Kontrastmittel) der Lungenregion um die Thymusdrüse
² Dekrement - Spezielle abnehmende Form einer Amplitude, aus der eine abnehmbare Muskelbelastbarkeit abgelesen werden kann. Das gegenteilige Inkrement, ist eine spezielle zunehmende Form einer Amplitude. Sollte sich dieses in einer Serienstimulation zeigen, ist neben der Myasthenia gravis, an ein LEMS (Lambert Eaton Myathenes Syndrom) differenzialdiagnostisch zu denken. Hier ist dann ein Test der Kalziumkanal-Antikörper (VGCC-AK) zu erwägen. Das LEMS ist wie die MG eine muskelschwächende Erkrankung.
Zur Labor-Diagnostik ist anzumerken, dass in 50 % der okulären und etwa 10–15 % der generalisierten Fälle keine AChR-AK und in der Hälfte dieser Fälle auch keine MuSK-AK oder andere Antikörper nachweisbar sind. Es handelt sich dann um eine seronegative MG. Auch hier ist die Diagnosestellung erschwert, und ist für myasthenie-erfahrene Neurologen, anhand des Verlaufs zu beurteilen.
Zu den elektrophysiologischen Untersuchungen ist zu sagen, dass gewisse Erfahrung zur Diagnostik notwendig sind. Zu Fehlerquellen gehören zu wenig untersuchte Muskeln, eine zu kalte Muskulatur und/oder ein durch den Untersucher zu wenig belasteter Muskel oder gar lediglich nur durch den Patienten während der Untersuchung betätigten Muskels sowie mangels Unwissen, z. B. zum rechtzeitigen Absetzen (falls medizinisch vertretbar) von beeinflussenden medikamentösen Therapien (z. B Pyridostigmin, den Cholinesterasehemmern. Auch kann erst ein im ersten Moment grenzwertiges Dekrement (7–9 %), nach erneuter Messung 2 und 5 Minuten posttetanisch, als pathologisch beurteilt werden.
Differenzial-Diagnostik
Gegenüber der Myasthenia Gravis sind vor allem, auszuschließen:
- Lambert-Eaton-Rooke-Syndrom LEMS
- Amyotrophe Lateralsklerose ALS
- Multiple Sklerose MS
- allgemeinere Muskelschwächen, verursacht durch z. B. Anorexie, Kachexie oder auch anderer seelischer, psychiatrischer oder neurologischer Erkrankungen
- Hirnerkrankungen, Durchblutungsstörungen, Stoffwechselstörungen
bzw. manifestieren sich abzugrenzende Sonderformen:
- neonatale Myasthenie (Neugeborene)
- medikamentös, boreolös bzw. durch Gifte erzeugte myasthene Symptome
- Myasthene Syndrome (kongenitale Myasthenie-Syndrome), die keine Autoimmunerkrankungen sind
Therapie
- Durch Cholinesterasehemmer, z. B. Neostigmin-Nasenspray oder Pyridostigmin-Tabletten bzw. Injektionslösungen (Kalymin®, Mestinon®), die den Abbau des Transmitters Acetylcholin (ACh) hemmen, wird die Konzentration des ACh im synaptischen Spalt erhöht und damit der Autoantikörper kompetitiv verdrängt.
- Immunsuppressive Behandlung mit Glucocorticoiden oder Azathioprin kann die Antikörperwirkung abgeschwächt werden. Bei schweren oder kritischen Verläufen werden eine Plasmapherese oder hochdosierte Immunglobulingaben eingesetzt.
- Chirurgische Entfernung des Thymus oder eines Thymoms (Thymektomie)
- Umstellung auf nicht-Lungen-aspirierende Kost, vorbeugende Schlucktherapie und in schweren Fällen Ernährung per PEG (Magensonde), apparativer Sekret-Absaugung und/oder Tracheostoma (→ Tracheotomie)
Verlauf und Prognose
Bleiben nur die Augenmuskeln betroffen, ist die Prognose gut und mit medikamentöser Therapie beherrschbar. Die Ausbreitung auf den gesamten Körper (Generalisierung) war hingegen vor einigen Jahren noch unheilbar. Durch moderne Medikamente ist in diesen Fällen die Prognose in den letzten Jahren deutlich besser geworden. Das bedeutet aber eine lebenslange Medikation. Weitere Maßnahmen, wie z.B. erwähnte PEG oder Tracheostoma wirken lebensverlängernd, können aber einen erheblichen Verlust an Lebensqualität bedeuten. Bei zu später oder gar fehlender Diagnosestellung, schweren Verläufen und Krisen (siehe Komplikationen) kann die Lebenszeit verkürzt sein.
Die Thymektomie führt vor allem bei jungen Patienten oft zu einem Rückgang der Symptome. In diesem Fall ist einige Jahre nach der Thymusentfernung evtl. auch das Absetzen der immunsuppressiven Medikamente möglich und somit eine deutliche Besserung, manchmal wird auch eine Remission erreicht.
Myasthenia gravis bei Tieren
Die Myasthenia gravis kommt vor allem bei Haushunden vor. Hier werden eine angeborene und eine erworbene Form unterschieden. Die Pathogenese ist die gleiche wie beim Menschen, also eine Autoimmunerkrankung, die sich gegen die Acetylcholinrezeptoren richtet. Die Behandlung erfolgt wie beim Menschen.
Angeborene Myasthenia gravis
Die angeborene Myasthenia gravis ist relativ selten und tritt gehäuft bei einigen Hunderassen (Jack Russell Terrier, Glatthaar Foxterrier, Springer-Spaniel) auf. Die Erkrankung wird autosomal-rezessiv vererbt und tritt typischerweise in der 6. bis 8. Lebenswoche auf.
Erworbene Myasthenia gravis
Die erworbene Myasthenia gravis ist häufiger und vermutlich multifaktoriell bedingt, wobei wohl ebenfalls eine genetische Prädisposition vorliegt. Thymome kommen ebenfalls als Auslöser in Betracht. Die Altersprädisposition ist zweiphasisch, mit einem Häufigkeitsmaximum bei Junghunden (1.-4. Lebensjahr) und einem bei alten Hunden (ab 9. Lebensjahr).
Klinisch äußert sie sich zumeist in lokalen Muskelstörungen. Häufig (etwa 80 % der Fälle) ist vor allem die Speiseröhre betroffen und es entwickelt sich ein Megaösophagus. Klinisch zeigt sich ein häufiges Regurgitieren. Hier besteht ein besonders hohes Risiko für eine Aspirationspneumonie. Auch die Rachenmuskulatur kann involviert sein, was sich in Stimmveränderungen äußert. Eine dritte häufige Lokalisation sind wie beim Menschen die Augenmuskeln. In diesem Fall ist der Lidschlussreflex vermindert und schnell ermüdbar. Die generalisierte Form äußert sich in schneller Ermüdbarkeit, steifem Gang bis hin zu einer Paraparese der Hintergliedmaßen oder Tetraparese. Auch ein Megaösophagus ist häufig. Eine myasthenische Krise mit akuter Para- oder Tetraparese und Megaösophagus kann ebenfalls auftreten.
Die Diagnose kann sicher nur mit dem Nachweis der Autoantikörper gestellt werden. Bei einem Megaösophagus oder bei Thymomen kann eine Röntgenaufnahme des Brustkorbs wichtige Hinweise geben. Differentialdiagnostisch müssen andere Polyneuropathien, Botulismus (selten beim Hund), Polymyositis, Zeckenparalyse (selten) und Azetylcholin- oder Organophosphat-Vergiftungen ausgeschlossen werden (Siehe auch VETAMIN D).
Literatur
- Publikationen der „Deutschen Myasthenie Gesellschaft“ e.V.
- Publikationen der „Deutschen Gesellschaft für Neurologie“ (Deutsche Gesellschaft für Neurologie)
- Publikationen der Fa. Temmler (pdf Zurück zur Bewegung)
- Hohenegger M: Allgemein-Anästhesie bei Myasthenia gravis; intensiv 2006; 14:72-75
- Wolfgang Köhler und Jörn P. Sieb: Myasthenia gravis (UNI-MED Science) - (Oktober 2003)
- Conti-Fine BM, Milani M, Kaminski HJ: Myasthenia gravis: past, present, and future. J Clin Invest. 2006 Nov;116(11):2843-54. PMID 17080188
- Juel VC, Massey JM: Myasthenia gravis. Orphanet J Rare Dis. 2007 Nov 6;2:44. PMID 17986328
Einzelnachweise
- ↑
- In „Neuromuscul Disorder“ UK.2005 Dec;15(12):878-86. Epub 2005 Nov 9, „The early history of myasthenia gravis“ von Hughes T. (Green College, University of Oxford)
- In „Neurol. Clinics“ 1994 May;12(2):231-42, „The history of myasthenia gravis.“ von Pascuzzi RM. (Department of Neurology, Indiana University School of Medicine, Indianapolis.)
- Publikationen der „Deutschen Myasthenie Gesellschaft“ e.V.
- ↑
- Publikationen der „Deutschen Myasthenie Gesellschaft“ e.V.
- Publikationen der „Deutschen Gesellschaft für Neurologie“ ([1] → MG)
- ↑
- Das EMG-Buch und periphere Neurologie in Frage und Antwort - Von Christian Bischoff, Wilhelm J. Schulte-Mattler, Bastian Conrad, Georg-Thieme-Verlag 2005 S. 288 ff
- Schluckstörungen - Diagnostik und Rehabilitation - David Buchholz, Gudrun Bartolome, Hubertus Feussner, Christian Hannig, Heidrun Schröter-Morasch, Stefanie Neumann, Christian Pehl, Mario Prosiegel, Anita Wuttge-Hannig - Mitwirkende Personen Gudrun Bartolome, Veröffentlicht von Elsevier GmbH Deutschland, 2006
- Publikationen der „Deutschen Myasthenie Gesellschaft“ e.V.
- uni-goettingen.de
- Publikationen der „Deutschen Gesellschaft für Neurologie“
Weblinks

Bitte beachte den Hinweis zu Gesundheitsthemen!

Wikimedia Foundation.