- Peronäale Muskelatrophie
-
Klassifikation nach ICD-10 G60.0 Hereditäre sensomotorische Neuropathie inkl. Charcot-Marie-Tooth-Hoffmann-Syndrom ICD-10 online (WHO-Version 2006) Der Morbus Charcot-Marie-Tooth (CMT) wurde nach ihren Entdeckern Jean-Martin Charcot (1825-1893), Pierre Marie (1853-1940) und Howard Tooth (1856-1926) benannt. Heute ist die Bezeichnung Hereditäre motorisch-sensible Neuropathie Typ I (HMSN I) üblicher. Sie wird auch Neurale Muskelatrophie genannt und gehört zu den neuromuskulären Erkrankungen.
Inhaltsverzeichnis
Epidemiologie
Die Charcot-Marie-Tooth-Krankheit ist die häufigste neurogenetische Erkrankung. 20–30 Personen auf 100.000 Einwohner sind betroffen. Es handelt sich in den meisten Fällen um ein autosomal dominant vererbtes Leiden, daher gibt es Häufungen innerhalb einzelner Familien. In den meisten Fällen wird die CMT durch eine Mutation auf dem Chromosom 17 hervorgerufen.
Ätiologie
CMT ist eine vererbliche Erkrankung der peripheren Nerven, bei der durch eine genetische Mutation entweder der Nervenzellfortsatz, Axon genannt, oder die isolierende Myelinschicht geschädigt wird. Das Myelin könnte mit einer Plastikhülle um ein Elektrokabel verglichen werden. Es wird die saltatorische Erregungsleitung, also die Fortleitung von Nervenimpulsen im peripheren Nerven behindert, so dass die Befehle des Gehirns die Muskeln nicht richtig erreichen können. Aus der Denervierung folgt eine Schwäche und ein Abbau der betroffenen Muskulatur.
Anders als bei den Muskeldystrophien werden die Menschen, die CMT haben, mit normalen Muskeln geboren. Die Muskeln schwinden (atrophieren), weil die von CMT beeinträchtigten Nerven die Befehle des Gehirns für bestimmte Muskelbewegungen nicht exakt übertragen können.
Exakt kommt es zu einer Duplikation des PMP ( Peripheral Myelin Protein) Gens auf Chromosom 17. Dies führt wiederum dazu, dass sich die Myelinscheide verdickt und die Versorgung mit Nährstoffen erschwert wird. Auf Dauer führt dies zu einer Schädigung der Myelinscheide bzw. des Nervs. Je stärker die Myelinschicht geschädigt ist, desto geringer ist die Nervenleitgeschwindigkeit und desto schwerwiegender die Ausprägung des Krankheitsbildes. Normal ist eine Nervenleitgeschwindigkeit von 50 m/s. Bei der CMT liegt eine Leitgeschwindigkeit von ca. 40 m/s vor.
Symptome
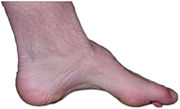 Hohlfuß mit Atrophie der kleinen Fußmuskeln und Hammerzehen bei einem Patienten mit CMT
Hohlfuß mit Atrophie der kleinen Fußmuskeln und Hammerzehen bei einem Patienten mit CMTDie Erkrankung setzt oft schon im Kindesalter ein. Manchmal kommt es aber auch erst zwischen dem 20. und dem 30. Lebensjahr zu ersten Manifestationen. Die wichtigsten Symptome dieser Krankheit bestehen in einer zunehmenden Schwäche von Händen und Füßen, die sich nach und nach auf die Arme und Beine ausbreitet. Manchmal bleibt den Betroffenen nur die Zuhilfenahme von Krücken oder sogar ein Leben im Rollstuhl.
In der Regel ist der Musculus tibialis anterior (Fußheber), der vorn am Schienbein herabläuft, als erster Muskel betroffen. Daraus resultiert ein unsicherer Gang: Der Fuß hängt schlaff herunter, man stolpert leicht und muss das Bein vom Oberschenkel aus anheben, bis auch die Zehen sich vom Boden lösen. Der Fuß setzt patschend wieder auf dem Boden auf. Dies wird anschaulich als Stepper- oder Storchengang bezeichnet.
Die Reflexe, besonders der Achillessehnenreflex, fallen frühzeitig aus.
Diagnose
Die Messung der (erheblich reduzierten) Nervenleitgeschwindigkeit und die Nervenbiopsie stützen die Diagnose. Inzwischen ist darüber hinaus eine genetische Untersuchung zur Identifikation der zugrundeliegenden Mutation möglich. Eine kausale Behandlung gibt es nicht.
Verlauf und Prognose
Bei längerem Verlauf ist die Atrophie der Unterschenkelmuskulatur auf Anhieb sichtbar, die Unterschenkel wirken grazil, während die Oberschenkelmuskulatur noch kräftig ausgebildet sein kann. Sensible Reizsymptome (Schmerzen, Missempfindungen, Muskelkrämpfe) gehören zum Krankheitsbild. Die motorischen Ausfallserscheinungen sind jedoch ausgeprägter und bestimmen das Beschwerdebild. Die Atrophie der Muskulatur schreitet annähernd symmetrisch voran. Insgesamt ist der Verlauf sehr langsam und dauert über Jahrzehnte an. Eine Heilung der genetisch bedingten Erkrankung ist bisher nicht möglich.
Literatur
- Auer-Grumbach M: Hereditary sensory neuropathy type I. Orphanet J Rare Dis. 2008 Mar 18;3:7. Review. PMID 18348718

Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.