- Muskeldystrophie
-
Muskeldystrophien, auch progressive Muskeldystrophie (Dystrophia musculorum progressiva), sind eine Gruppe von Muskelerkrankungen. Es handelt sich um Erbkrankheiten, die durch Mutationen im Erbgut verursacht werden, welche meist zu Defekten oder zu einem Mangel von in der Muskulatur vorkommenden Proteinen führen. Dies führt in der Konsequenz zu Muskelschwäche und Muskelschwund. Alle Muskeldystrophien sind durch fortschreitende (progressive) Degeneration der Muskulatur, einhergehend mit Umbauprozessen, gekennzeichnet. Diese Veränderungen werden zusammengefasst als dystrophische Veränderungen, die sich licht- oder elektronenmikroskopisch nachweisen lassen. Die einzelnen Muskeldystrophien unterscheiden sich hinsichtlich der Art des Erbgangs, der hauptsächlich betroffenen Körperregionen, des Erkrankungsalters und des Verlaufs. Eine kausale Behandlungsmöglichkeit, die das Fortschreiten der Muskeldegeneration aufhalten kann, ist nicht bekannt.
Inhaltsverzeichnis
Einteilung
Klassifikation nach ICD-10 G71.0 Muskeldystrophie ICD-10 online (WHO-Version 2011) Die Einteilung der Muskeldystrophien kann entweder traditionell nach klinischem Verteilungsmuster, dass heißt anhand der bevorzugt betroffenen Muskulatur, oder nach genetischen Kriterien erfolgen. Eine einheitliche Klassifikation, die sich in der Fachliteratur durchgesetzt hat, gibt es noch nicht.
Gruppe Erbkrankheit Vererbung OMIM Genort Genprodukt X-Chromosomal vererbte Dystrophien Muskeldystrophie Typ Duchenne X-chromosomal-rezessiv 310200 Xp21 Dystrophin Muskeldystrophie Typ Becker X-chromosomal-rezessiv 300376 Xp21 Dystrophin Emery-Dreifuss-Muskeldystrophie Typ 1 X-chromosomal-rezessiv 310300 Xq28 Emerin Skapuloperoneale Muskeldystrophie X-chromosomal-rezessiv 300696 Xq26.3 Four and a half LIM domain protein 1 (FHL1) Reducing body myopathy (RBM) X-chromosomal-rezessiv 300717 und 300718 Xq26.3 Four and a half LIM domain protein 1 (FHL1) Gliedergürteldystrophien LGMD1A autosomal-dominant 159000 5q31 Myotilin LGMD1B autosomal-dominant 159001 1q11-21 Laminin A/C LGMD1C autosomal-dominant 60781 3p25 Caveolin-3 LGMD1D autosomal-dominant 602067 6q23 nicht bekannt LGMD1E autosomal-dominant 603511 7q nicht bekannt LGMD2A autosomal-rezessiv 253600 15q15.1-21.1 Calpain-3 LGMD2B autosomal-rezessiv 253601 2p13 Dysferlin LGMD2C autosomal-rezessiv 253700 13q12 γ-Sarkoglykan LGMD2D autosomal-rezessiv 608009 17q12-21.3 α-Sarkoglykan (Adhalin) LGMD2E autosomal-rezessiv 604286 4q12 β-Sarkoglykan LGMD2F autosomal-rezessiv 601287 5q33-34 δ-Sarkoglykan LGMD2G autosomal-rezessiv 601954 17q11-12 Telethonin LGMD2H autosomal-rezessiv 254110 9q31-33 E3 Ubiquitin-Ligase (TRIM32) LGMD2I autosomal-rezessiv 607155 19q13 Fukutin-related protein (FKRP) LGMD2J autosomal-rezessiv 608807 2q31 Titin LGMD2K autosomal-rezessiv 609308 9q34.13 POMT1 LGMD2L autosomal-rezessiv 611307 11p14.3 Anoctamin-5 LGMD2M autosomal-rezessiv 611588 9q31-33 Fukutin LGMD2N autosomal-rezessiv 613158 1p34 POMT2 LGMD2O autosomal-rezessiv 613157 1p34.1 POMGNT1 LGMD2Q autosomal-rezessiv 613723 8q24.3 Plectin Kongenitale Muskeldystrophien Kongenitaler Merosinmangel (Merosinopathie, MDC1A) autosomal-rezessiv 607855 6q22-23 Laminin-α2 (Merosin) Kongenitale Myopathie mit Integrin-alpha-7-Mangel autosomal-rezessiv 613204 12q13 Integrin alpha-7 Kongenitale Muskeldystrophie Typ 1C (MDC1C) autosomal-rezessiv 606612 19q13 Fukutin-related protein (FKRP) Kongenitale Muskeldystrophie Typ Fukuyama (FCMD) autosomal-rezessiv 253800 9q31-33 Fukutin Walker-Warburg-Syndrom autosomal-rezessiv 236670 9q34.13 POMT1 Muskel-Auge-Gehirn-Krankheit (Typ Santavuori) autosomal-rezessiv 253280 1p34.1 POMGNT1 Rigid Spine-Syndrom autosomal-rezessiv 602771 1p36.11 Selenoprotein N1 Ullrich-Myopathie autosomal-rezessiv und autosomal-dominant 254090 21q22.3
21q22.3
2q37.3Kollagen alpha-1
Kollagen alpha-2
Kollagen alpha-3Bethlem-Myopathie autosomal-dominant 158810 21q22.3
21q22.3
2q37.3Kollagen alpha-1
Kollagen alpha-2
Kollagen alpha-3Distale Muskeldystrophien Distale Muskeldystrophie Typ Welander autosomal-dominant 604454 2p13 nicht bekannt Distale Muskeldystrophie Typ Udd autosomal-dominant 600334 2q31.2 Titin Distale Muskeldystrophie Typ Markesbery-Griggs autosomal-dominant 609452 10q23.2 ZASP Distale Muskeldystrophie Typ Nonaka autosomal-rezessiv 605820 9p13.3 UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine-Kinase (GNE) Muskeldystrophie Miyoshi 1 autosomal-rezessiv 254130 2p13.2 Dysferlin Muskeldystrophie Miyoshi 2 autosomal-rezessiv 613318 8q22.3 unbekannt Muskeldystrophie Miyoshi 3 autosomal-rezessiv 613319 11p14.3 Anoctamin-5 Distale Muskeldystrophie Typ Laing autosomal-dominant 160500 14q11.2 Myosin-7 Vocal cord and pharyngeal weakness with distal myopathy (VCPDM) autosomal-dominant 606070 5q31 Matrin-3 Myofibrilläre Myopathien Myofibrilläre Myopathie Typ 1 (MFM1) autosomal-dominant und autosomal-rezessiv 601419 2q35 Desmin Myofibrilläre Myopathie Typ 2 (MFM2) autosomal-dominant 608810 11q23.1 Alpha-crystallin B Myofibrilläre Myopathie Typ 3 (MFM3) autosomal-dominant 609200 5q31.2 Myotilin Myofibrilläre Myopathie Typ 4 (MFM4) autosomal-dominant 609452 10q23.2 ZASP Myofibrilläre Myopathie Typ 5 (MFM5) autosomal-dominant 609524 7q32.1 Filamin-C Myofibrilläre Myopathie Typ 6 (MFM6) autosomal-dominant 612954 10q26.11 BAG family molecular chaperone regulator 3 (BAG3) Myotone Dystrophien Myotone Dystrophie Typ 1 (Morbus Curschmann-Steinert) autosomal-dominant 160900 19q13.32 Myotonin-Protein-Kinase (DMPK, Myotonic dystrophy protein kinase) Myotone Dystrophie Typ 2 (Proximale myotone Myopathie, PROMM) autosomal-dominant 602668 3q21.3 Zink finger protein 9 (ZNF9) Sonstige Muskeldystrophien Fazioskapulohumerale Muskeldystrophie (FSHD) autosomal-dominant 606070 4q35 Double homeobox protein 4 (DUX4) Skapuloperoneale Muskeldystrophie (Stark-Kaeser-Syndrom) autosomal-dominant 181400 2q35 Desmin Emery-Dreifuss-Muskeldystrophie Typ 4 autosomal-dominant 612998 6q25.1-q25.2 Nesprin-1 Emery-Dreifuss-Muskeldystrophie Typ 5 autosomal-dominant 612999 14q23.2 Nesprin-2 Genetische Klassifikation der Muskeldystrophien[1][2] Dystrophinopathien
Die Muskeldystrophie des Typs Duchenne ist die häufigste muskuläre Erbkrankheit im Kindesalter (1 : 5000). Sie beginnt im Kleinkindalter mit einer Schwäche der Becken- und Oberschenkelmuskulatur, schreitet rasch voran und endet, meist im jungen Erwachsenenalter, immer tödlich, sobald die Herz- und Atemmuskulatur abgebaut wird. Man bezeichnet sie daher auch als „maligne (= bösartige) Muskeldystrophie“.
Die Muskeldystrophie des Typs Becker-Kiener ist wesentlich seltener als jene des Typs Duchenne (1 : 60000) und betrifft ebenfalls bevorzugt die Becken- und Oberschenkelmuskulatur. Die Erkrankung tritt im Schulalter auf und schreitet langsamer voran. Aufgrund des wesentlich günstigeren Krankheitsverlaufes bezeichnet man diesen Typ als „benigne (= gutartige) Muskeldystrophie“.
Ursachen
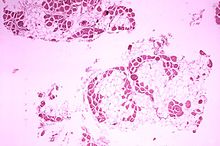 Histopathologisches Bild eines Querschnitts durch den Wadenmuskel eines Patienten mit Muskeldystrophie Typ Duchenne. Die Muskelfasern (rot) sind ausgedehnt durch Fettzellen (optisch leer = weiß) ersetzt.
Histopathologisches Bild eines Querschnitts durch den Wadenmuskel eines Patienten mit Muskeldystrophie Typ Duchenne. Die Muskelfasern (rot) sind ausgedehnt durch Fettzellen (optisch leer = weiß) ersetzt.
Bei den Dystrophinopathien wird ein für die Stabilität der Muskelmembran wichtiges Protein, das Dystrophin, gar nicht oder in funktionsgestörter Form gebildet, was früher oder später zum Untergang von Muskelfasern und Ersatz durch Fett- oder Bindegewebe führt.
Bei der Muskeldystrophie Typ Duchenne ist eine Mutation (60–70 % Deletion, ca. 5 % Duplikation, ca. 35 % Punktmutation) im Dystrophin-Gen (Locus Xp21.2) nachgewiesen. Mehr als 98 % der Mutationen verursachen hierbei eine Verschiebung (Frameshift) des Leserasters und führen damit zu einem kompletten Verlust des Dystrophinproteins. Dabei sind etwa 1/3 der Fälle Neumutationen und nur 2/3 von den Eltern direkt vererbt.
Bei der Muskeldystrophie Typ Becker ist eine besondere Art der Mutation („in-frame“) des Dystrophin-Gens für die Erkrankung verantwortlich. Dabei entsteht meist – im Gegensatz zum Typ Duchenne – ein in seiner Funktion stark beeinträchtigtes Dystrophin. Der Typ Becker-Kiener ist in seiner Häufigkeit seltener (ca. 1 auf 20.000) als Typ Duchenne (ca. 1 auf 3.500) und zeigt durch das Vorhandensein eines „Restproteins“ meist ein nicht so schweres Krankheitsbild.
Symptomatik und Verlauf
Die Muskeldystrophie vom Typ Duchenne wurde von Guillaume-Benjamin Duchenne bereits im 19. Jahrhundert in Paris beschrieben. Im 3. bis 5. Lebensjahr fällt bei den Betroffenen eine leichte Muskelschwäche der Beine auf, die zu häufigem Stolpern und Fallen führt. Im weiteren Verlauf ist das Treppensteigen nur mit Zuhilfenahme eines Geländers möglich. Die Muskelschwäche der Becken- und Oberschenkelmuskulatur verursacht einen Watschelgang sowie ein erschwertes Aufstehen aus dem Sitzen oder Liegen. Die Kinder klettern an sich selbst hoch (Gowers-Manöver) oder nutzen Wände und Möbel zum Abstützen. Ab dem 5. bis 7. Lebensjahr sind das Treppensteigen und Aufstehen aus dem Sitzen oder Liegen nur noch mit Hilfe anderer möglich, da die Erkrankung auch auf die Muskulatur der Schulter und Arme übergreift. Zwischen dem 7. und 12. Lebensjahr ist ein Anheben der Arme in die Waagerechte kaum mehr möglich. Viele Kinder sind in diesem Alter bereits auf den Rollstuhl angewiesen, können sich aber noch eingeschränkt selbständig versorgen.Meist besteht ab dem 18. Lebensjahr vollständige Pflegebedürftigkeit.
Infolge des Muskelschwundes kommt es zu schmerzhaften Fehlstellungen von Gelenken und Knochenverformungen. Charakteristisch für den Typ Duchenne sind die sogenannten „Gnomen-“ oder „Kugelwaden“. Sie entstehen durch Fetteinlagerungen in der bindegewebig umgebauten Unterschenkel-Muskulatur („Pseudohypertrophie“). Bei Abbau der Schultergürtelmuskulatur kommt es zu hervorstehenden Schulterblättern („Scapulae alatae“), auch Engelsflügel genannt. Durch Schwäche der Atemmuskulatur wird das Abhusten, bei Infekten der Luftwege, deutlich erschwert und dadurch kann die Lebenserwartung erheblich eingeschränkt werden. Der Herzmuskel ist zwar meist vom Krankheitsprozess betroffen, doch führen erhöhte Herzfrequenz und sonstige Veränderungen des Rhythmus oder auch Beeinträchtigung der Herzkraft selten zu subjektiven Beschwerden. Die Lebenserwartung der Patienten beträgt je nach Verlauf bis zu 40 Jahre, jedoch versterben einzelne Patienten auch schon vor Beginn der Pubertät.
Die Muskeldystrophie vom Typ Becker-Kiener nimmt einen langsameren Verlauf. Auch hier ist zuerst die Becken- und Oberschenkelmuskulatur betroffen. Der Erkrankungsbeginn ist sehr variabel (2. bis 35. Lebensjahr) überwiegend jedoch zwischen dem 6. und 18. Lebensjahr. Die Gehfähigkeit der Patienten bleibt meist bis zum 40., gelegentlich sogar bis zum 60. Lebensjahr erhalten. Trotz des insgesamt günstigeren Verlaufs versterben auch hier die meisten Patienten in Folge einer zunehmenden Herzschwäche oder wegen Komplikationen im Rahmen der Atemschwäche. Bedeutsam ist, dass sich die Herzschwäche auch unabhängig vom Schweregrad der Schwäche der Körpermuskulatur entwickeln kann.
Diagnose
Der Verdacht auf Muskeldystrophie ergibt sich, wenn im Kindesalter eine ungewöhnliche, symmetrisch ausgebildete Schwäche der Muskulatur beobachtet wird. Bei der Anamnese interessieren besonders der Beginn der Funktionsstörungen, der Verlauf und das Auftreten ähnlicher Störungen in der Familie. Bei der körperlichen Untersuchung wird nach allgemeinen Auffälligkeiten geschaut, wie Haltung, Beweglichkeit und Atmung. Bei der neurologischen Untersuchung wird die Funktion der Nerven und Muskeln überprüft. Die Gendiagnose macht eine genaue Zuordnung nach Typus möglich.
Muskelschwäche kann neurogene oder myogene Ursachen haben. Die Differenzierung erfolgt durch Elektroneurographie und Elektromyographie. Bei der Elektroneurographie werden die Nervenleitgeschwindigkeit und die Funktionstüchtigkeit der Nervenfasern ermittelt. Dabei werden Nerven elektrisch gereizt. Bei der Elektromyographie werden Muskelströme gemessen. Dabei kann ermittelt werden, ob die Nervenfasern geschädigt oder ob die Muskeln selbst erkrankt sind.
Laborwerte geben ebenfalls wichtige Hinweise: Die Menge an Creatin-Kinase, einem Muskelenzym, ist oft im Blut erhöht. Andere Enzyme und Stoffwechselprodukte der Muskeln, bestimmte Antikörper, sowie andere Blut- und Urinwerte ergänzen die Diagnose. Als bildgebende Verfahren werden MRT und Ultraschall eingesetzt. Hier können ohne Belastung des Patienten strukturelle Veränderungen der Muskeln beurteilt werden. Die Muskelbiopsie erlaubt die Untersuchung des Muskels unter dem Licht- oder Elektronenmikroskop. Die Stoffwechselvorgänge im Muskel können damit genau untersucht werden.
Therapie
Eine effektive Behandlung bedarf einer Kombination aus Arzt, Physiotherapie, Ergotherapie, Pflege. Kortisongaben und operative Lösung von Kontrakturen verlängern die Gehfähigkeit, Kreatin kann eine diskrete Verbesserung des Muskelenergiestoffwechsel bewirken (regelmäßige Therapiepausen nötig). Eine sich entwickelnde Skoliose (Verkrümmung der Wirbelsäule) kann durch die Implantation von Metallstäben (Harrington-Stab) operativ begradigt werden; hierdurch wird vor allem die Atemfunktion verbessert. Seit 1989 werden mitwachsende Teleskopstäbe eingesetzt. Als Hilfsmittel werden Orthesen, Rollstuhl, Duschstuhl, Badewannenlifter, Toilettensitzerhöhungen und Rollstuhlrampen eingesetzt. Da durch die zunehmende Schwäche der Atemmuskulatur die Eigenatmung immer weiter abnimmt, kann eine maschinelle Beatmung notwendig werden. Dies erfolgt überwiegend nicht-invasiv, über eine Nasen- oder Nasen-Mund-Maske. Die Durchführung einer Tracheotomie ist meist erst spät nötig, wenn der Patient auch tagsüber auf die Beatmung angewiesen ist. Besonders zu beachten ist auch die Abhustschwäche. Durch physiotherapeutische Techniken oder apparative Maßnahmen ist auch dabei eine Unterstützung möglich. Eine mit der Muskeldystrophie einhergehende Fatigue kann – neben hinreichenden Ruhephasen – mit Vitamininfusionen und Sauerstoffbehandlungen positiv beeinflusst werden. Ein weiterer Ansatz beruht darauf, das defekte Gen trotz der Mutation vollständig auszulesen. Dies soll z. B. mit Hilfe von Ataluren möglich sein.
Ein Versuch, die Duchenne-Muskeldystrophie gentherapeutisch zu behandeln, schlug fehl. Grund dafür war vermutlich eine Autoimmunreaktion nach dem Versuch, die entsprechenden Gene in ein Muskelprotein einzubringen. Weitere Studien mit adjuvanter Immunmodulation wurden im Oktober 2010 durchgeführt.[3]
Ziel aller physiotherapeutischen Maßnahmen ist es, die Folgen der Muskelschwäche so gering wie möglich zu halten bzw. in einem bestimmten Ausmaß zu korrigieren. Die Behandlung orientiert sich an Typ, Stadium und Prognose der Erkrankung, sowie an der Lebenssituation des Betroffenen und seiner Familie.
Allgemeine Schwerpunkte sind
- Erhalten einer ausreichenden Atemkapazität
- Erhalten / Ökonomisieren der Herz-Kreislauf-Funktion
- Erhalten / Verbessern der Durchblutung
- Erhalten der Beweglichkeit / Kontrakturprophylaxe
- Kräftigen / Ökonomisieren der intakten Muskulatur
- Erhalten der Koordination
- Erhalten der Orthostatik (Aufrichtung des Körpers)
- DTKP-Prophylaxe (Dekubitus, Thrombose, Kontrakturen, Pneumonie)
- Verzögerung von Skoliosen (Wirbelsäulenverbiegung)
- Erhalten der Selbständigkeit
Physiotherapie wird vielfach von den Patienten als angenehm und hilfreich empfunden. So ist schon die gezielte Zuwendung zu dem betroffenen Muskel, mit dem etwas geschieht, hoffnunggebend. Zu bedenken ist aber, dass jedenfalls die Dystrophinopathien auf einer geminderten Stabilität der Muskelmembran gegen mechanische Kräfte beruhen. Jede zusätzliche Bewegung führt daher vermutlich zu einer weiteren Schädigung des Muskels, in der Summe und nach Jahren dann zur Schwäche. Die Versuchung für alle – Physiotherapeut und Betroffene – ist groß, durch „Muskelaufbau“ die Schwäche zu kompensieren („Nach der Reha ging alles besser“), dafür aber langfristig den Verlauf zu beschleunigen. Auch gibt es keinen gesicherten Nachweis der Wirksamkeit bestimmter physiotherapeutischer Maßnahmen.
Die Auswahl und die Stärke der Behandlungsmaßnahmen muss aufgrund der progressiven Muskelschwächung ständig neu bewertet werden. Generell sollte nur mit warmer Muskulatur gearbeitet werden (vorherige Wärmeanwendung / Zimmertemperatur um 30°). Da dystrophe Muskulatur durch Überanstrengung und Überforderung zusätzlich geschädigt wird, sollte der Therapeut auf das Arbeiten gegen Widerstand bzw. mit Gewichten und Aufprallbelastungen (Springen) verzichten. Bei Kindern soll die Spontanaktivität und das Erlernen eines ökonomischen Krafteinsatzes angeregt werden (z. B. Bewegungsübergänge mit Abstützen). Bei allen Angeboten sollten die Kinder Freude an der Therapie haben. Spaß an der Bewegung kann helfen, bestehende Depressionen zu lindern.
Die Muskelschwäche ist das Hauptproblem bei Muskeldystrophien. Bei rasch fortschreitenden Erkrankungen besteht das Risiko einer verstärkenden Abschwächung durch zu intensives Training. Es sollte grundsätzlich mehr mit dem Ziel der Funktionsbesserung als mit dem Ziel der Kräftigung trainiert werden. Darum müssen Patienten über die Warnzeichen einer Überbelastung informiert sein. Hierzu gehören ein Schwächegefühl innerhalb von 30 Minuten nach der Übung oder Muskelschmerzen 24 bis 48 Stunden nach dem Training. Andere Warnsignale beinhalten ausgeprägte Muskelkrämpfe, Schweregefühl von Armen und Beinen und anhaltende Kurzatmigkeit. Dennoch ist durch ökonomisches Training mit leichter bis mäßiger Belastung wie Gehen, Schwimmen und Fahren auf dem Ergometer eine Verbesserung der Ausdauer und damit eine Minderung der Schwäche zu erreichen.
Sobald die Krankheit auf die Atemmuskulatur übergreift, müssen regelmäßige Lungenfunktionstests erfolgen. In den letzten Jahren wurden gute Fortschritte in der Möglichkeit der Heimbeatmung gemacht. Bei der Erstellung eines Trainingsprogramms muss die Herzbelastbarkeit überprüft werden. Manchmal kann der Einsatz eines Herzschrittmachers notwendig sein.
Behandlungsmaßnahmen:
- Atemtherapie
- Wärmeanwendungen
- Passives Durchbewegen
- Traktionen / Gleitmobilisation
- Kombination aus isometrischen und dynamischen Bewegen
- Stemmführungen nach Brunkow
- Lagewechsel und Transfer einüben
- intermittierendes Ausdauertraining
- Klopf-Druck-Massage
- Klassische Massage (nur sanfte Griffe)
- Ultraschall-Therapie
- Elektrotherapie (Galvanisation)
- Hydroelektrische Bäder
- Ergometertraining
- Schwimmen / Bewegungsbad
Kontraindikationen: Zu vermeiden ist/sind
- Arbeiten an kalter Muskulatur
- Kältereize
- Training bis zur Ermüdung / Erschöpfung
- Dehnung der betroffenen Muskulatur
- Reizstromtherapie
- kräftige Massagereize (Knetungen, tiefe Friktionen)
- Resistives Bewegen der dystrophen Muskulatur
Diagnostik
Die Bestimmung der Kreatinkinase (CK) im Blutserum kann einen allgemeinen Anhalt für das Vorliegen einer Muskeldystrophie bieten, da die Serumkonzentration bei Degeneration der Skelettmuskelfasern ansteigt. Dementsprechend ist die Kreatinkinase bei den meisten Muskeldystrophien leicht bis deutlich erhöht (HyperCKämie). Die Bestimmung der Serumkreatininkinase gilt als sensitiver und spezifischer als die Bestimmung anderer Enzyme wie der Aspartat-Aminotransferase (ASAT), der Alanin-Aminotransferase (ALAT) und der Lactatdehydrogenase (LDH), die häufig ebenfalls erhöht sind.[4] Da sich die Höhe der Serumkreatininkinase zwischen den einzelnen Muskeldystrophien teils deutlich unterscheidet, kann diese auch für differentialdiagnostische Überlegungen herangezogen werden. Darüber hinaus kann eine CK-Erhöhung auch einziges Symptom einer Muskeldystrophie sein. Dies spielt u. a. eine Rolle bei der Untersuchung von Verwandten, da eine Erhöhung der CK dann gegebenenfalls Hinweise auf den Vererbungsmodus liefern kann.[5]
Siehe auch
- Muskeldystrophie-Netzwerk – interdisziplinäres Netzwerk zur Erforschung von Muskeldystrophien
Weblinks
- Deutsche Gesellschaft für Muskelkranke e. V. (DGM)
- Schweizerische Gesellschaft für Muskelkranke SGMK
Einzelnachweise
- ↑ Robert Griggs: Muscular Dystrophies: 101 (Handbook of Clinical Neurology Revised Series). Elsevier Science 24. Mai 2011, S. 2-3, ISBN 978-0080450315
- ↑ OMIM, Aufruf am 1. September 2011
- ↑ New England Journal of Medicine 2010; 363:1429-1437
- ↑ Anthony A. Amato, Robert C. Griggs: Overview of the muscular dystrophies. In: Robert Griggs: Muscular Dystrophies. 3. Auflage. Elsevier, 2011, ISBN 978-0-08-045031-5, S. 4 (Handbook of Clinical Neurology, Band 101)
- ↑ Dieter Pongratz, Stephan Zierz: Neuromuskuläre Erkrankungen. Diagnostik, interdisziplinäre Therapie und Selbsthilfe Deutscher Ärzteverlag, 2003, ISBN 978-3769111729, S. 73

Bitte den Hinweis zu Gesundheitsthemen beachten!
Wikimedia Foundation.