- Systemischer Lupus erythematodes
-
Klassifikation nach ICD-10 M32.- Systemischer Lupus erythematodes M32.0 Arzneimittelinduzierter systemischer Lupus erythematodes M32.1+ Systemischer Lupus erythematodes mit Beteiligung von Organen oder Organsystemen L93.- Lupus erythematodes L93.0 Diskoider Lupus erythematodes L93.1 Subakuter Lupus erythematodes cutaneus ICD-10 online (WHO-Version 2006) Der Lupus erythematodes (lat. lupus „Wolf“, griech. ἐρυθήμα „Röte“) ist eine systemische Autoimmunerkrankung aus der Gruppe der Kollagenosen. Der Name „Lupus“ – „Wolf“ leitet sich von der Möglichkeit von Verstümmelungen im Gesichtsbereich durch CDLE-Läsionen her [1], Ärzte früherer Generationen verglichen diese Läsionen mit Wolfsbissen. Heutzutage kommt es dazu dank moderner Behandlungsmöglichkeiten nur noch selten. „Erythematodes“ (engl. erythematosus) – „errötend“ leitet sich von den bei dieser Krankheit häufig (bei 71 % der Patienten) vorkommenden Rötungen her. Besonders charakteristisch für SLE (systemischer Lupus erythematodes) ist das so genannte Schmetterlingserythem.
Neun von zehn Betroffenen sind weiblich (geschlechtsspezifischer Signalweg der Interferon-alpha-Induktion).[2] Siehe auch: Gynäkotropie. Schätzungen zufolge leiden in Deutschland etwa 40.000 Menschen, vor allem junge Frauen im gebärfähigen Alter, an Lupus erythematodes, während die Krankheit in den USA häufiger ist.
Inhaltsverzeichnis
- 1 Formen und Symptome
- 2 Ursache
- 3 Untersuchung
- 4 Behandlung
- 5 Unverträglichkeiten
- 6 Epidemiologie
- 7 Geschichte
- 8 Siehe auch
- 9 Literatur
- 10 Weblinks
Formen und Symptome
Systemischer Lupus erythematodes (SLE)
Der systemische Lupus erythematodes (SLE, andere Bezeichnung: Lupus erythematosus disseminatus, abgekürzt LED) beginnt oft mit Fieber; Abgeschlagenheit und Empfindlichkeit gegenüber Sonnenlicht folgen häufig. Außerdem klagen die Betroffenen meist über rheuma-ähnliche Gelenkschmerzen. Auf der Haut bilden sich oft Erytheme.
Einen Überblick über mögliche Symptome bei systemischem Lupus erythematodes gibt folgende Tabelle:
Mögliche SLE-Symptome [3] Häufigkeit in % Symptom 85 Gelenkschmerzen 84 Allgemeinbeschwerden
(Müdigkeit, Leistungsschwäche …)81 Hautveränderungen 77 Nierenbefunde 63 Gelenkentzündung 58 Raynaud-Syndrom 54 Beschwerden d.
Zentralnervensystem54 Schleimhautveränderungen 47 Magen-Darm Beschwerden 37 Rippenfellentzündung 32 Lymphknotenerkrankung 29 Herzbeutelentzündung 17 Lungenbeteiligung 5 Muskelentzündung 4 Herzmuskelentzündung 4 Bauchspeicheldrüsenentzündung Es kann auch jedes andere Körperorgan befallen werden (Systemischer = alle „Systeme“). Es können Entzündungen der Gelenke, von Herz, Lungen, Nieren und Hirn entstehen. Die Gefahr dabei liegt insbesondere im Multi-Organsystemversagen.
Der SLE kann akut-entzündlich mit schweren Schüben, foudroyant verlaufen. Diese Verlaufsform führte früher häufig und schnell durch Befall von Muskeln, Gelenken, Hirn, Haut und Nieren zum Tod. Sie ist heute dank moderner Behandlungsmethoden sehr selten geworden. Meist verläuft der SLE heute milder und mit weniger Krankheitszeichen. Es gibt langsam-schleichende und schubweise Verlaufsformen.
Es gibt auch sehr milde Verlaufsformen, bei denen nur ein oder zwei ACR-Kriterien erfüllt sind [1]. Diese Verlaufsformen können nach jahrelangem Verlauf in einen SLE übergehen.
SLE mit sekundärem Antiphospholipid-Syndrom (APS)
Bei manchen Formen (ca. 40 % der Betroffenen) des Lupus erythematodes kommt es zur Bildung von Antikörpern gegen Phospholipid-Protein-Komplexe (anti-Cardiolipin und anti-beta-2-Glykoprotein I Antikörper als Hauptvertreter), was man sich bei der Diagnostik zunutze macht, da diese in immunologischen Tests nachgewiesen werden können. Außerdem können diese Komplexe zu Thrombembolien führen und so einen schlechten Krankheitsverlauf prognostizieren lassen. Weibliche Patienten mit APS haben ein erhöhtes Risiko für wiederkehrende Fehlgeburten (ein Merkmal bei APS-Patientinnen).
Medikamenteninduzierter SLE
Die mildere Verlaufsform des LE, ausgelöst durch Antihypertensiva (etwa Dihydralazin), Antiarrhythmika (etwa Procainamid), Antikonvulsiva und Antibiotika, aber auch andere Medikamente, bessert sich meist nach Absetzen des Medikaments. Laborparameter: ANA und Histon-AK.
SLE im Spätstadium
Da das späte Krankheitsstadium heute medikamentös fast immer vermieden werden kann, tritt das Vollbild des SLE nur noch sehr selten auf. Die typischen Organschäden durch den akuten SLE kommen daher nur noch in der Regel abgemildert vor. Heutzutage wird durch Medikamente versucht, diese Schäden zu vermeiden oder zu verringern; viele Patienten erreichen daher das späte, früher oft letal ausgehende Krankheitsstadium nicht. Neben einer regelmäßigen Kontrolle durch einen versierten Rheumatologen ist auch die medikamentöse Behandlung unbedingt wichtig. Die möglichen, durch SLE ausgelösten, schweren Organschäden Niereninsuffizienz, nephrotisches Syndrom, Arteriosklerose, Herzklappenschäden, Thromboembolien, Knochennekrosen, neurologische und psychiatrische Erkrankungen, Lungenfibrose [1] sind dadurch in der heutigen Zeit seltener geworden.
Chronisch-diskoider Lupus erythematodes (CDLE)
Beim Hautlupus (CDLE) ist nur die Haut betroffen. Der Verlauf ist milde. Nur bei etwa 5 % entwickelt sich ein SLE. Der Hautlupus manifestiert sich meistens in scheibenförmiger Form („diskoider“ Lupus erythematodes). Diese Hautveränderungen zeigen einen dreiphasigen Aufbau:
- Außen besteht eine Rötung, die in einen
- schuppenden Bereich übergeht. Die Hautschuppen sitzen fest. Nach Entfernung einer Schuppe findet man an deren Unterseite einen sog. keratotischen Sporn (Tapeziernagel-Phänomen).
- Innen zeigt sich ein Gewebsschwund (Atrophie), der vernarbt und im Haarbereich zum dauerhaften Haarausfall (Alopezie) führt. siehe Bild bei DermIS UV-Licht und bestimmte Medikamente provozieren die Hautveränderungen
Hypertropher CDLE
Tumider Lupus erythematodes
Der Lupus erythematodes tumidus ist eine seltene Variante des diskoiden Lupus erythematodes mit entzündlichen tumorförmigen Infiltraten, die oft im Gesicht entstehen. Die rötlichen Herde sind geschwollen, überwärmt und haben durch Vernarbungen eine gesprenkelte Oberfläche.
Subakut kutaner Lupus erythematodes (SCLE)
Seltene Sonderform des Hautlupus, die in einen SLE übergehen kann.
Neonatales LE-Syndrom
Im Falle einer mütterlichen Autoimmunerkrankung wie dem systemischen Lupus erythematodes ist das Risiko für eine Fehlgeburt bis auf etwa 20 % erhöht. Von den übrigen 80 % entwickelt nur ein geringer Anteil einen neonatalen Lupus erythematodes. Grundsätzlich können die Autoantikörper der Mutter ab der 16. Schwangerschaftswoche über den Mutterkuchen auf das Kind übertragen werden. Hier rufen sie bevorzugt Symptome an der Haut und am Herzen hervor. Die Mutter selbst kann zu diesem Zeitpunkt durchaus noch symptomlos sein. In einer prospektiven Studie wurden Hautveränderungen in 16 %, ein AV-Block °III in 1,6 %, erhöhte Leberwerte in 26 % und hämatologische Auffälligkeiten in 27 % der Fälle beschrieben.[4] Symptome an anderen Organen wie den Nieren, den Lungen, dem Nervensystem oder den Blutzellen treten sehr viel seltener auf. Der AV-Block ist auch durch eine immunsuppressive Therapie nicht wieder rückgängig zu machen und muss meist mit einem Schrittmacher behandelt werden. Ansonsten hat die Erkrankung eine gute Prognose, weil die übrigen Symptome mit Abbau der passiv von der Mutter übertragenen Autoantikörper von alleine wieder verschwinden. Da längst nicht alle Kinder von Müttern mit den entsprechenden Autoantikörpern auch Symptome entwickeln, ist eine prophylaktische immunsuppressive Therapie nicht grundsätzlich indiziert. Allerdings wird empfohlen, in solchen Fällen ab der 16. Schwangerschaftswoche regelmäßig Ultraschalluntersuchungen des Herzen des Feten durchzuführen. Wird hierbei ein AV-Block °I oder II gefunden, wird die prophylaktische Behandlung der Mutter mit einem Steroid diskutiert.[5]
Ursache
Die Ätiologie des SLE ist weitgehend unbekannt. Man geht allerdings davon aus, dass Viren oder UV-Licht als ursächliche Faktoren in Frage kommen. Es kommt zur Apoptose von Zellen, sodass deren Kernbestandteile (Autoantigene) freigesetzt werden. Dies gilt insbesondere dann, wenn die Phagozytose der apoptotischen Zellen gestört ist, wie es für eine Subgruppe der SLE-Patienten beschrieben ist (Herrmann M, Voll RE, et al.: Arthritis and Rheumatism. 1998 41(7):1241–50). Diese Zellen werden durch das fehlgesteuerte Immunsystem als fremd erkannt, und es werden Antikörper gegen sie gebildet, sogenannte Autoantikörper. Autoantigene und Autoantikörper bilden Immunkomplexe. Diese können vom mononukleären phagozytären System (Makrophagen) nicht beseitigt werden und lagern sich an den verschiedensten Orten (etwa an der Gefäßwand) ab. Es kommt zur Komplementaktivierung, Thrombozytenaktivierung …, und eine Gefäßentzündung (Vaskulitis) entsteht. Daraus entstehen ein Gefäßverschluss und später die Organläsion. Die Kernbestandteile werden jedoch auch auf der Oberfläche der verschiedenen Zellen präsentiert (etwa von Keratinozyten). Das fehlregulierte Immunsystem reagiert darauf, und es kommt zur polyklonalen B-Zell-Aktivierung und zur Produktion von weiteren Autoantikörpern, was dann zur Zerstörung der Zellen führt (s. Schmetterlingserythem).
Die Ursache für diese krankhafte Reaktion des Immunsystems ist bisher unbekannt. Es gibt eine starke genetische Disposition. Familienangehörige ersten und zweiten Grades zeigen eine erhöhte SLE-Prävalenz (Peter & Pichler, Klinische Immunologie, 1996, S. 348). Die Wahrscheinlichkeit, dass ein SLE-Patient mindestens einen Verwandten ersten Grades mit SLE hat, wird mit 3 bis 10 Prozent angegeben (Miehle, Rheumatologie in Praxis und Klinik, 2000, S. 911). Für eineiige Zwillinge wird eine gemeinsame SLE-Erkrankung von 57 % berichtet, für zweieiige Zwillinge von 5 % (Peter & Pichler, s.o.). Auch weitere Autoimmunkrankheiten kommen im Verwandtenkreis gehäuft vor.
Clearance Defizienz als ein mögliches Entstehungsmodel für SLE
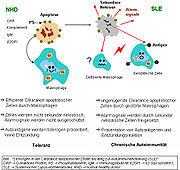 Ineffiziente Clearance
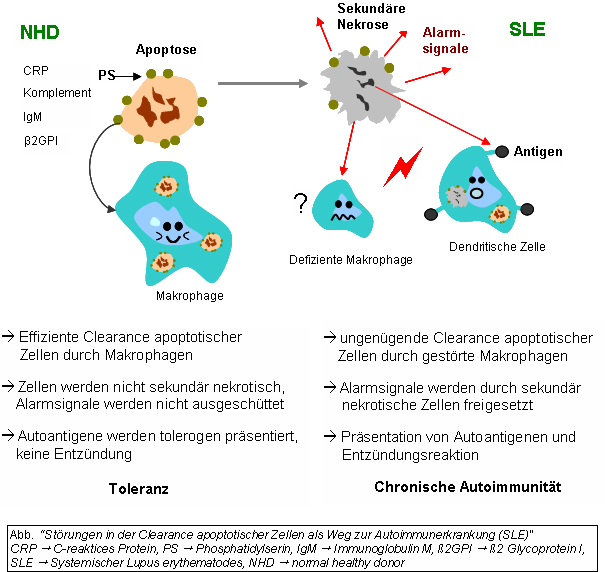
Ineffiziente ClearanceDer präzise Entstehungsmechanismus des Systemischen Lupus erythematodes (SLE) ist ein multifaktorielles Geschehen und noch unklar. Neben den bereits erwähnten Ursachen ist die gestörte Clearance von sterbenden Zellen ein weiterer Grund zur Ausprägung dieser Autoimmunerkrankung. Dies bezieht sich, neben der gesteigerten Apoptoseanfälligkeit, auf die gestörte Phagozytoseaktivität von Monozyten oder Gewebsmakrophagen und Granulozyten sowie auf fehlende oder ineffiziente Serumkomponenten.
Isolierte Monozyten aus peripherem Venenblut von SLE-Patienten zeigen eine verminderte Ausprägung des CD44 Oberflächenmoleküls, das am Phagozytose-Prozess beteiligt ist. Die meisten dieser Monozyten sowie einige der „Sternhimmel-Makrophagen“ (tingible body macrophages, TBM), die in den Keimzentren der Lymphknoten zu finden sind, zeigen außerdem ganz klar einen morphologischen Unterschied bei SLE-Patienten: Sie sind nicht nur vermindert und sterben eher, sondern sie sind auch kleiner und fehlgebildet. Serumkomponenten, wie Komplementfaktoren, CRP und einige Glykoproteine, sind außerdem ausschlaggebend für den funktionierenden Phagozytose-Prozess. Solche Komponenten fehlen oder sind im Serum vieler SLE-Patienten vermindert oder ineffizient.
Die Clearance von früh apoptotischen Zellen ist eine wichtige Funktion im Organismus. Ist diese Fähigkeit gestört und kommt es zum Fortschreiten der Apoptose-Phasen, so kommt es zur sekundären Nekrose der Zelle. Von nekrotischen Zellen, die ihre Membranintegrität verloren haben, werden Kernbestandteile als Autoantigene sowie „danger signals“ präsentiert, womit die Reifung dendritischer Zellen (DC) stimuliert wird. Eine „ineffiziente Clearance“ wird auch durch ein hohes Aufkommen apoptotischer Zellen ausgelöst. Eine erhöhte Apoptose-Rate löst somit auch die Reifung der DC aus, sowie die Präsentation der intrazellulären Antigene spät-apoptotischer Zellen oder sekundär-nekrotischer Zellen über deren MHC-Moleküle.
Autoimmunität kann u.a. durch eine verlängerte Exposition intrazellulärer und nukleärer Autoantigene spät-apoptotischer und sekundär-nekrotischer Zellen initiiert werden. Die B- und T-Zell-Toleranz gegenüber Autoantigenen apoptotischer Zellen geht verloren, und die Lymphozyten werden durch Autoantigene aktiviert. Die Entzündungsreaktion und die Antikörperproduktion durch Plasmazellen resultieren als typisches Krankheitsbild des SLE. Bei Patienten mit Chronisch-diskoidem Lupus erythematodes (CDLE / Hautlupus) wurde dieser Clearance-Defekt ebenfalls festgestellt.
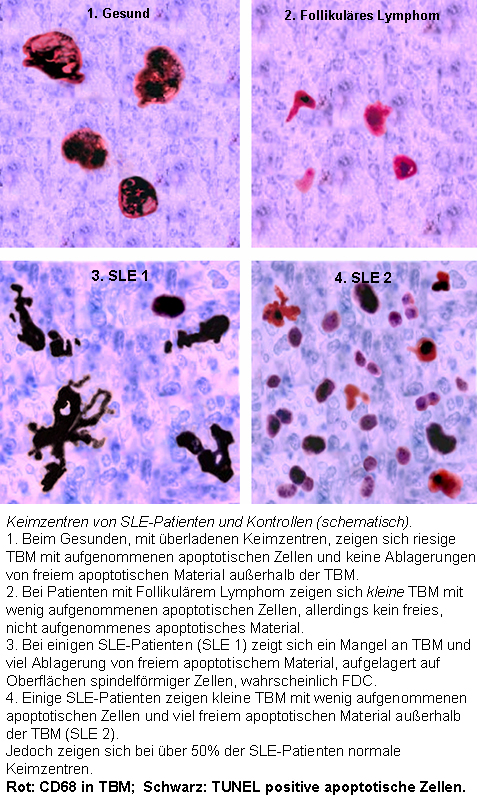
 Keimzentren in SLE und Kontrollen
Keimzentren in SLE und KontrollenAkkumulation in Keimzentren
Beim Gesunden werden apoptotische Lymphozyten in den Keimzentren der Lymphknoten durch spezialisierte Phagozyten, TBM, effizient abgeräumt, wodurch kein freies apoptotisches und potentiell autoantigenes Material dort vorkommt. Bei SLE-Patienten kommt es dort, aufgrund der ineffizienten Clearance, zur Ablagerung und Anhäufung apoptotischen Materials. In enger Nachbarschaft zu den TBM sind, in dem besonderen Mikromilieu des Keimzentrums, follikuläre dendritische Zellen (FDC) lokalisiert, die im Gegensatz zu den im Knochenmark gereiften dendritischen Zellen (DC) die Antigene auf ihrer Oberfläche binden und sie weder aufnehmen, noch über MHC-Moleküle präsentieren. Autoreaktive B-Lymphozyten gehen, wie nicht-autoreaktive B-Zellen, aus der somatischen Hypermutation von Zentroblasten hervor und wandern in die helle Zone ein. Die durch Zufall gebildeten autoreaktiven B-Lymphozyten erhalten normalerweise keine Überlebenssignale durch FDC und sterben durch Apoptose. Ist die Clearance ineffizient, so kommt es zur Ansammlung apoptotischen Kernmaterials in der hellen Zone der Keimzentren, das durch FDC fixiert wird. Die FDC, die mit abgelagerten Kern-Autoantigenen beladen sind, können Kurzzeit-Überlebenssignale für autoreaktive B-Lymphozyten darstellen. Finale Überlebenssignale, die die Differenzierung zu autoantikörperproduzierenden Plasmazellen und zu B-Gedächtniszellen zulassen, erhalten die autoreaktiven B-Zellen durch Interaktion mit autoreaktiven T-Helfer-Zellen nach dem Einwandern in die Mantelzone. Die chronische Autoimmunerkrankung SLE kann die Folge sein.
Untersuchung
CDLE (Hautlupus)
Neben dem typischen Hautbild wird oft eine kleine Hautprobe entnommen (Biopsie) und feingeweblich untersucht, um die Diagnosestellung zu sichern. Bei der Untersuchung unter dem Mikroskop zeigen sich in den betroffenen Hautarealen eine Hyperkeratose, Verstärkung der Basalmembran und eine hohe Anzahl von CD4-T-Lymphozyten. Im Rahmen einer Spezialuntersuchung werden fluoreszierende Antikörper eingesetzt, die sich an bestimmte Bereiche der Hautprobe binden und ein typisches Bandenmuster bilden (positiver Lupusband-Test).
SLE (systemischer LE)
Die Diagnose systemischer Lupus erythematodes richtet sich nach den sogenannten ARA-Kriterien (ARA steht für: American Rheumatology Association – 1988 in American College of Rheumatology (ACR) umgenannt). Bei Vorhandensein von 4 der 11 Kriterien kann mit 80- bis 90-prozentiger Sicherheit die Diagnose eines SLE gestellt werden.
ACR- Kriterien (vormals ARA-Kriterien)
Die 11 ACR-Kriterien (1982, revidiert 1997) lauten [1]:
- Schmetterlingserythem: Darunter versteht man eine umschriebene, symmetrische, hellrote, flache bis leicht erhabene Rötung der Wangen, die über dem Nasenrücken zusammenläuft.
- CDLE- typische Hautveränderungen
- Photosensibilität (Überempfindlichkeit gegenüber Licht: Nach Sonneneinstrahlung können beispielsweise Kopfschmerzen, Abgeschlagenheit und Fieber auftreten – dies hat in diesem Zusammenhang nichts mit Sonnenbrand oder Sonnenstich zu tun!)
- Erosionen oder Geschwüre der Mundschleimhaut
- Gelenksschmerzen und Gelenkserguss
- Serositis (Entzündung von sogenannten serösen Häuten wie dem Lungenfell oder dem Herzbeutel)
- Nierenbefall:
- Mehr als 0,5 g Eiweiße pro Tag im Urin oder
- krankhafte Harnsedimente wie Blutreste oder Harnzylinder
- Befall des Zentralnervensystems:
- epileptische Anfälle oder
- psychische Krankheiten ohne bekannte metabolische oder medikamentöse Ursache als Auslöser
- hämatologische Symptome:
- Hämolytische Anämie oder
- Leukopenie mit Leukozytenzahlen unter 4000 pro mm³ Blut oder
- Lymphopenie mit Lymphozytenzahlen unter 1500 pro mm³ Blut oder
- Thrombozytopenie mit Thrombozytenzahlen unter 100000 pro mm³ Blut
- Immunologische Befunde:
- Positives LE-Zellzeichen: In Blutausstrichen sichtbare „hematoxylin bodies“: Darunter versteht man phagozytierte Kernreste in Leukozyten. Diese Kernreste stammen von zerstörten Zellen, an die ein bestimmter, gegen Desoxyribonukleoproteine gerichteter Autoantikörper, der sogenannte „LE-Faktor“, gebunden hatte. Die Untersuchung auf das LE-Zellzeichen wird heutzutage kaum mehr durchgeführt und ist nurmehr von historischem Interesse [1]; oder
- Autoantikörper gegen native DNA oder
- Autoantikörper gegen das Ribonukleoprotein Sm
- Autoantikörper gegen Phospholipide
- Antinukleäre Antikörper (ANA) in der Immunfluoreszenz-Mikroskopie
Behandlung
Allgemeines
Regelmäßige Untersuchungen sind erforderlich, um frühzeitig den Befall von Organen wie etwa Niere oder Herz erkennen zu können. Da UV-Licht die Hauterscheinungen des Lupus erythematodes verschlimmert, ist konsequenter Sonnenschutz notwendig.
Medikamente
Die medikamentöse Therapie des Lupus ist stufenförmig aufgebaut und richtet sich nach der Stärke der Beschwerden, der Organbeteiligung und der Wirksamkeit bei den einzelnen Patienten.
Bei leichter Erkrankung ist in einigen Fällen gar keine oder nur eine symptomatische Behandlung mit Hautcremes und/oder sogenannten NSAR (nichtsteroidalen Antirheumatika) nötig.
Die nächste Stufe ist zumeist das Basistherapeutikum Chloroquin (den DMARDs zugehörig), das auch zur Behandlung von Malaria angewendet wird, häufig kombiniert mit Cortison.
Erreicht man mit diesen Mitteln keine Verbesserung des Zustandes, werden zumeist immunsuppressive Medikamente eingesetzt. Das häufigste und in der Behandlung von Lupus am besten erprobte Medikament ist Azathioprin. Auch mit Cyclosporin A und Mycophenolat-Mofetil werden Erfolge erzielt. Außerdem kommen gelegentlich Zytostatika wie Cyclophosphamid oder Methotrexat zum Einsatz. Auch Thalidomid wird unter strenger Überwachung eingesetzt. Mitunter werden einzelne dieser Medikamente untereinander kombiniert, häufiger ist die Kombination mit Cortison.
Andere Behandlungsmethoden
Andere Behandlungsverfahren sind Immunadsorption, Plasmapherese, Stammzelltransplantation, Immunglobuline und physikalische Therapie.
Empfängnisverhütung
Lange Zeit galt die Einnahme der „Pille“ (orale Empfängnisverhütung – Kontrazeption) bei Patientinnen mit Lupus erythematodes als gefährlich, da man befürchtete, die in der Pille enthaltenen Östrogene könnten zu einem Aufflackern oder zur Verschlimmerung der Erkrankung führen.
Andererseits gibt es gute Gründe für die Anwendung der „Pille“ gerade bei Patientinnen mit Lupus erythematodes:
- Geplante Schwangerschaften haben in einer Phase geringer Krankheitsaktivität (Remission) weniger Komplikationen.
- Patienten mit hoher Krankheitsaktivität oder Patientinnen, die fruchtschädigende (teratogene) Medikamente einnehmen müssen, benötigen eine zuverlässige Verhütungsmethode.
- Bei Patientinnen, die Glukokortikoide einnehmen müssen, kann die Einnahme der „Pille“ den damit verbundenen Knochenschwund vermindern und so einer Osteoporose vorbeugen.
- Die Anwendung der Spirale bei gleichzeitiger immunsuppressiver Therapie schützt nicht zuverlässig vor Schwangerschaften und verschleiert unter Umständen Entzündungen im Genitaltrakt.
2005 konnten Petri et al.[6] sowie Sánchez-Guerrero et al.[7] [8] zeigen, dass bei Patientinnen mit inaktiver Erkrankung oder moderater, aber stabiler Krankheitsaktivität die Einnahme von hormonellen Kombinationspräparaten sicher ist, wenn keine Phospholipid-Antikörper nachweisbar sind. Patientinnen mit schwerer Erkrankung, Störungen der Blutgerinnung oder Phospholipid-Antikörpern im Blut sollten die Pille hingegen nicht einnehmen.
Unverträglichkeiten
Allgemein bekannt sind die Unverträglichkeiten auf Carotin (insbesondere in Karotten) und Anisol (insbesondere in Anis, Kümmel und Fenchel).
Epidemiologie
In Europa tritt der systemische Lupus erythematodes mit einer Inzidenz von 25 bis 27 jährlich Neuerkrankten pro 100.000 Personen auf.
Beim SLE liegt die 5-Jahresüberlebensrate bei 95 %, die 10-Jahresüberlebensrate bei 85 %.
Geschichte
Erstbeschreiber der Erkrankung war P.L.A. Cazenave (1851).
Siehe auch
Lupus pernio, Lupus vulgaris, Antiphospholipidsyndrom, CREST-Syndrom
Literatur
- ↑ a b c d e Peter Fritsch: „Dermatologie und Venerologie“, Springer Verlag, 2. Auflage 2004, ISBN 3-540-00332-0
- ↑ Bein,G.;Hackstein,H.: Warum ist Lupus Erythematodes eine Frauenkrankheit? In: Biol Unserer Zeit 2007;3(27): 154–155“
- ↑ Hettenkofer, Hans-Jürgen [Hg.]: Rheumatologie. Stuttgart: Thieme Verlag, 1998, S. 91
- ↑ R. Cimaz, D. L. Spence, L. Hornberger et al.: Incidence and spectrum of neonatal lupus erythematosus: a prospective study of infants born to mothers with anti-Ro autoantibodies. In: J Pediatr 2003;142: 678–683
- ↑ V. Wahn: Neonataler Lupus erythematodes. In: Monatsschrift Kinderheilkunde 2006, 154:1203–1206
- ↑ Petri M, Kim MY, Kalunian KC, et al. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med 2005;353:2550–2558
- ↑ Sánchez-Guerrero J, Uribe AG, Jiménez-Santana L, et al. A trial of contraceptive methods in women with systemic lupus erythematosus. N Engl J Med 2005;353:2539–2549.
- ↑ Sánchez-Guerrero J, Uribe AG, Jiménez-Santana L, et al. A trial of contraceptive methods in women with systemic lupus erythematosus. N Engl J Med 2005;353:2539–2549.„One patient receiving combined oral contraceptives died from amoxicillin-related severe neutropenia“
Weblinks
- Bilder der Lupus-Hautveränderungen bei DermIS
- Engl. Informationen zu SLE (U.S. National Library of Medicine und NIH)
- Manson JJ, Rahman A: Systemic lupus erythematosus. Orphanet J Rare Dis. 2006 Mar 27;1:6. PMID 16722594

Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.