- Zuker-Algorithmus
-
Der Zuker-Algorithmus berechnet die optimale Sekundärstruktur einer RNA-Sequenz mit der minimalen freien Energie unter einem gegebenen thermodynamischen Modell. Es ist also ein Algorithmus zur RNA-Strukturvorhersage. Der Algorithmus verwendet die Methode der Dynamischen Programmierung und wurde 1981 veröffentlicht. Das RNA-Strukturvorhersageprogramm mfold implementiert eine veränderte Version dieses Algorithmus.
Inhaltsverzeichnis
Idee
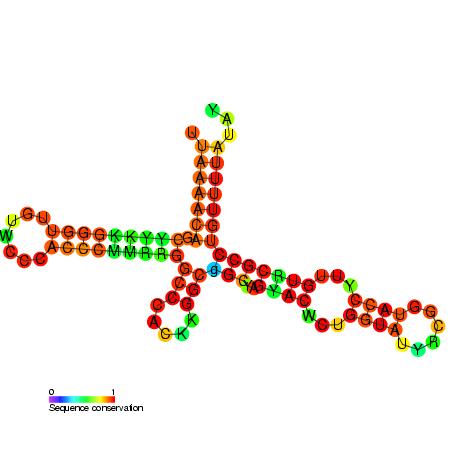
 Kleeblattartige Sekundärstruktur der Konsensus-Sequenz eines CIS-Element der Enterovirus-Familie.
Kleeblattartige Sekundärstruktur der Konsensus-Sequenz eines CIS-Element der Enterovirus-Familie.
Eine gegebene RNA-Sequenz kann aufgrund vieler möglicher Kombinationen von Basenpaarungen in exponentiell viele verschiedene Sekundärstrukturen gefaltet werden. Bei der Strukturvorhersage muss über dem Suchraum nach einem bestimmten Kriterium optimiert werden. Beispielsweise kann die Struktur mit den meisten Basenpaarungen ausgewählt werden. Diese Struktur kann allerdings biologisch bzw. biochemisch unrealistisch sein, da sie z. B. eine Hairpinloop mit nur einer ungepaarten Base enthält oder eine energetisch sehr instabile Struktur darstellt.
Ein biologisch sinnvolleres Kriterium ist die Betrachtung der freien Energie einer Sekundärstruktur. Ist die freie Energie einer Struktur kleiner als die freie Energie einer anderen Struktur, dann ist die erste Struktur stabiler als die zweite. Unter Laborbedingungen kann die freie Energie von vielfältigen Teilstrukturen einer RNA-Sekundärstruktur gemessen werden. Ein Beispiel für so eine Datensammlung ist [1]. Aus diesen Daten kann dann die freie Energie einer gegebenen RNA-Sekundärstruktur einer gegebenen RNA-Sequenz approximiert werden.
Der Algorithmus berechnet nun bei einer gegebenen RNA-Sequenz die Struktur mit der minimalen freien Energie. Die unter diesem Modell optimalen Sekundärstrukturen werden von Experten (Biologen, Biochemikern) als biologisch realistischer beurteilt.
Algorithmus
Die RNA-Sequenz wird mit s bezeichnet, wobei n = | s | . Die minimale freie Energie einer optimalen Sekundärstruktur wird rekursiv für alle Teilsequenzen von s berechnet. Die Matrix W speichert in der Zelle W[i,j] die minimale freie Energie (mfe) für die Teilsequenz s[i..j] von s. Die Matrix V speichert in der Zelle V[i,j] die mfe der Struktur der Teilsequenz s[i..j], wobei s[i] und s[j] ein Basenpaar sind.
Initialisierung: W[i,j] = 0,j − i = 4
V[i,j] = 8.4kcal / mol,j − i = 4
Kurze RNA-Moleküle formen keine stabile Sekundärstruktur.
Rekursion:
![W[i,j] = \min\begin{Bmatrix}
W[i+1,j] \\
W[i,j-1] \\
\min_{i<k<j-1} \{ W[i,k] + W[k+1,j] \} \\
V[i,j]
\end{Bmatrix}, i<j, j-i><span class=](c/25c5a342aa01a2952852289eb5f69f49.png) 4, 0
4, 0Die Fallunterscheidung berücksichtigt vier Fälle. Im ersten bzw. zweiten Fall setzt sich die optimale Struktur für s[i..j] aus dem optimalen Struktur der Teilsequenz s[i + 1,j] bzw. s[i,j − 1] und einer ungepaarten Base links bzw. rechts davon zusammen. In beiden Fällen ändert sich nichts an der mfe.
Im dritten Fall wird die optimale Struktur für s[i..j] gebildet, in dem die optimalen Strukturen der geteilten Sequenz konkateniert werden. Die mfe ist die Summe der mfe der Teilstrukturen der Teilsequenzen s[i,k] bzw. s[k + 1,j].
Der 4. Fall ermittelt die mfe für eine Teilsequenz von s[i..j], falls die Basen s[i] und s[j] ein Basenpaar darstellen.
Falls s[i] nicht mit s[j] ein Basenpaar bilden kann, dann wird
V[i,j] = inf
gesetzt.
Ansonsten wird V[i,j] wie folgt berechnet:
![V[i,j] = \min\begin{Bmatrix}
fh(i,j) \\
\min_{i<a<b<j} \{ fl(i,j,a,b) + V(a,b) \} \\
\min_{i+1<k<j-2} \{ W(i+1,k) + W(k+1, j-1) \}
\end{Bmatrix}, i<j, j-i><span class=](2/da2f159015bb3ef8fcb8dafc95085ed2.png) 4, 0
4, 0Die Funktion fh gibt die freie Energie für einen Hairpin-Loop der Teilsequenz s[i..j] zurück. Beispielsweise können die Werte für verschiedene Loop-Größen und Basenpaare experimentell unter einheitlichen Bedingungen ermittelt werden und sind in einer Hilfstabelle gespeichert. Die Funktion fl ist ein Stellvertreter und liefert die freie Energie für eine Internal-Loop, eine Bulge-Loop oder eine Stacking-Region, an der die Teilsequenzen s[i..a] s[j..b] beteiligt sind. Die mfe ist in diesem Fall die Summe dieses Konstruktes und der mfe für die Teilsequenz s[a,b], welche von einem Basenpaar eingeschlossen sein muss. Der vierte Fall modelliert eine Verzweigung der rekursiv konstruierten Struktur.
Backtracing: In der Zelle W[0,n − 1] ist nach der Berechnung der Matrix-Rekurrenzen die mfe für die gesamte RNA-Sequenz unter einer optimalen Sekundärstruktur gespeichert. Um eine optimale Sekundärstruktur zu ermitteln, welche die mfe hat, muss Optimierung via Backtracking zurückverfolgt werden.
Komplexität
Die beiden Tabellen W und V sind quadratisch, also liegt der Speicherbedarf in O(n2). Für jede Zelle muss – bei trivialer Implementierung – über O(n2) Möglichkeiten optimiert werden, denn interior loops benötigen 2 Variable. Durch ein geschicktes Preprocessing, also ein Vorab-Berechnen der Werte für interior loops kann man aber auch diesen Energiebeitrag in linearer Zeit bestimmen. Alternativ kann man die Größe eines loops mit einer Konstante beschränken. Damit liegt die Laufzeit des Algorithmus in O(n3).
Fallunterscheidung
Die Fallunterscheidung in der Rekursion der W-Rekurrenz des Algorithmus lässt sich auch kompakter formulieren, wenn die Sekundärstrukuren als Vienna-Strings (Dot-Bracket-Strings) abgebildet werden. Wenn ein Punkt eine ungepaarte Base und eine Klammer eine gepaarte Base bezeichnet, dann entspricht die Fallunterscheidung in der Rekursion von W folgender Fallunterscheidung
u = v. | .v | (v) | vw,
wobei u die Gesamtsekundärstruktur eines Teilstrings bezeichnet und v bzw. w Teilstrukturen von u bezeichnen.
Diese Beschreibung ist äquivalent zu folgenden grafischen Darstellung:
-
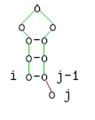
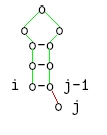
j ungepaart
-
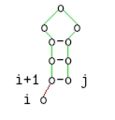
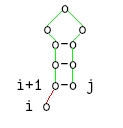
i ungepaart
-
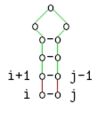
i und j gepaart
-
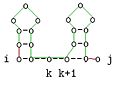
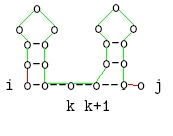
Verzweigung
Backtracing
Beim Backtracing können aufgrund der Fallunterscheidung des Zuker-Algorithmus mehrere unterschiedliche Backtracing-Pfade die gleiche Sekundärstruktur repräsentieren. Die Fallunterscheidung ist semantisch mehrdeutig. Beispielsweise eine Rekursion in W von Fall 1 nach Fall 2 nach Fall 1 erzeugt die gleiche Struktur wie die Rekursion von Fall 1 nach Fall 1 nach Fall 2. Für ein weiteres Beispiel siehe [2].
Diese Mehrdeutigkeit ist nicht problematisch bei der Ausgabe einer optimalen Sekundärstruktur. Wenn allerdings alle co-optimalen Sekundärstrukturen ausgegeben oder gezählt oder alle suboptimalen Strukturen bis zu einer gewissen Schranke ausgegeben oder gezählt werden sollen, dann ist das Backtracing zumindest nur noch schwer fehlerfrei, effizient und vollständig implementiertbar. Bei einer Standard-Backtracing-Implementation werden die redundanten Strukturen in exponentieller Anzahl ausgegeben bzw. gezählt.
Abgrenzung
Der Algorithmus verwendet eine weitere Tabelle im Vergleich zu dem Nussinov-Algorithmus, um eine Folge von gepaarten Basen (also eine Stacking-Region), unterschiedlich bewerten zu können. Eine ähnliches Muster verwendet auch der Gotoh-Algorithmus zur Berechnung des paarweisen Sequenzalignment bei affinen Gapkosten.
Der Nussinov-Algorithmus von 1978 berechnet die Sekundärstruktur einer RNA-Eingabesequenz, welche die maximale Anzahl von Basenpaaren hat. Da diese Strukturen nicht unbedingt biologisch sinnvoll sind, ist der Nussinov-Algorithmus nicht praxisrelevant. In der Praxis werden unter anderem RNA-Sekundärstrukturvorhersage Algorithmen verwendet, die die Struktur mit der minimalen freien Energie berechnen bzw. die Strukturen mit den kleinsten freien Energien bis zu einer gewissen Schwelle bestimmen. Die Verwendung des Zuker-Algorithmus in der mfold-Implementation ist immer noch verbreitet, obwohl inzwischen auch Algorithmen zur Sekundärstrukturvorhersage existieren, die eine detaillierte Fallunterscheidung vornehmen und deren Fallunterscheidung nicht mehrdeutig ist. Ein Beispiel für so einen verbesserten mfe-Algorithmus ist der Wuchty98-Algorithmus.
Literatur
- Michael Zuker and Patrick Stiegler: Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. In: Nucleic Acids Research. 9, Nr. 1, 1981, S. 133-148.
- Durbin et al.: Biological sequence analysis. Cambridge, 2006, ISBN 0-521-62971-3, S. 274-276.
- Jens Reeder: Algorithms for RNA secondary structure analysis : prediction of pseudoknots and the consensus shapes approach. 2007, S. 13-15 (http://bieson.ub.uni-bielefeld.de/volltexte/2008/1276/).
Quellen
-

Wikimedia Foundation.