- Enzephalopathie durch GLUT1-Defekt
-
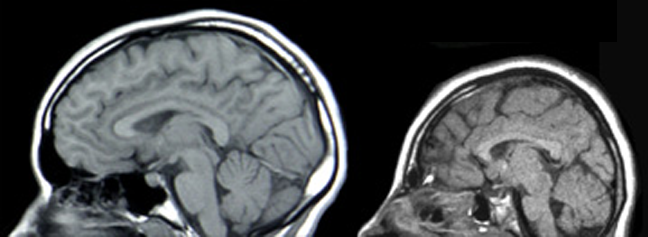
Klassifikation nach ICD-10 G93.4 Enzephalopathie, nicht näher bezeichnet ICD-10 online (WHO-Version 2006)  Kernspintomographische Aufnahme eines normalen Schädels (links) und einer Mikrozephalie (rechts). Die Mikrozephalie ist in diesem Fall durch eine Mutation des ASPM-Gens bedingt.
Kernspintomographische Aufnahme eines normalen Schädels (links) und einer Mikrozephalie (rechts). Die Mikrozephalie ist in diesem Fall durch eine Mutation des ASPM-Gens bedingt.Das GLUT1-Defizit-Syndrom, auch als Enzephalopathie durch GLUT1-Defekt bezeichnet, ist eine äußerst seltene, autosomal-dominant[1] vererbte Krankheit.
Inhaltsverzeichnis
Ätiologie, Genetik und Krankheitsverlauf
Die Ursache der Erkrankung ist eine Mutation des SLC2A1-Gens auf Chromosom 1 Genlocus p35-p31.3. Es handelt sich dabei meist um Neumutationen. Das Genprodukt GLUT1 ist ein aus 492 Aminosäuren bestehendes Membranprotein. GLUT1 ist ein wichtiger Membrantransporter für Glucose, ein Glucosetransporter. Die Bandbreite an Mutationen ist dabei hoch. So wurden bei den betroffenen Patienten große Deletionen, kleine Deletionen, Nonsense-Mutationen und Missense-Mutationen festgestellt.[2]
Das GLUT1-Defizit-Syndrom wird autosomal-dominant vererbt. Im Fall der Vererbung hat meist ein Elternteil eine leichte Form des Syndroms. Die Krankheit manifestiert sich neonatal beziehungsweise im Kleinkindalter und wird durch einen Mangel an GLUT1-Transportern im Endothel der Blut-Hirn-Schranke hervorgerufen. Dadurch wird das Gehirn nicht ausreichend mit D-Glucose versorgt und die betroffenen Patienten zeigen unter anderem eine Mikrozephalie, psychomotorische Retardierungen, Ataxie und eine Reihe anderer neurologischer Störungen.[3] Der Phänotyp ist jedoch sehr unterschiedlich und es wurden bisher verschiedene atypische Varianten beschrieben.[4]
Die GLUT1-Defizit-Syndrom ist eine äußerst seltene Erbkrankheit. Von 1991, dem Jahr der Erstbeschreibung[3], bis 2002 wurden weniger als 100 Patientien mit dieser Krankheit beschrieben.[5]
Therapie
Das GLUT1-Defizit-Syndrom ist derzeit nicht heilbar. Mit Hilfe einer ketogenen Diät lassen sich die Symptome gut kontrollieren oder zumindest lindern.[6] Die Diät sollte so früh wie möglich begonnen und bis in die Pubertät aufrechterhalten werden.[7] Während Erwachsene etwa 20 % der gesamten im Körper befindlichen Glucose im Gehirn verbrauchen, liegt bei Kindern der Anteil bei bis zu 80 %.[5] Deshalb kann nach der Pubertät die Diät normalerweise beendet werden.
Einzelnachweise
- ↑ J. Klepper u. a.: Autosomal dominant transmission of GLUT1 deficiency. In: Hum Molec Genet 10, 2001, S. 63–68. PMID 11136715
- ↑ D. Wang u. a.: Mutational analysis of GLUT1 (SLC2A1) in Glut-1 deficiency syndrome. In: Hum Mutat 16, 2000, S. 224–231. PMID 10980529
- ↑ a b D. C. De Vivo u. a.: Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. In: NEJM 325, 1991, S. 703–709. PMID 1714544
- ↑ J. Klepper: Glucose transporter deficiency syndrome (GLUT1DS) and the ketogenic diet. In: Epilepsia 49, 2008, S. 46–49. PMID 19049586
- ↑ a b J. Klepper und T. Voit: Facilitated glucose transporter protein type 1 (GLUT1) deficiency syndrome: impaired glucose transport into brain - a review. In: Eur J Pediatr 161, 2002, S. 295–304. PMID 12029447 (Review)
- ↑ D. Wang u. a.: Glut-1 deficiency syndrome: clinical, genetic, and therapeutic aspects. In: Ann Neurol 57, 2005, S. 111–118. PMID 15622525
- ↑ J. Klepper und B. Leiendecker: GLUT1 deficiency syndrome--2007 update. In: Dev Med Child Neurol 49, 2007, S. 707–716. PMID 17718830 (Review)
Literatur
- K. Brockman u. a.: Autosomal dominant glut-1 deficiency syndrome and familial epilepsy. In: Ann Neurol 50, 2001, S. 476-485. PMID 11603379
Weblinks
- GLUT1-Defizit-Syndrom bei Online Mendelian Inheritance in Man
- GLUT1-Defizit-Syndrom bei Orphanet (Datenbank für seltene Krankheiten)

Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.