- Schrankenstörung
-
Störungen der Blut-Hirn-Schranke können durch eine Reihe von verschiedenen Erkrankungen hervorgerufen werden. Die Blut-Hirn-Schranke kann aber auch selbst der Ausgangspunkt für einige sehr seltene neurologische Erkrankungen sein.
Inhaltsverzeichnis
- 1 Unmittelbar mit der Blut-Hirn-Schranke assoziierte Erkrankungen
- 2 Mittelbar mit der Blut-Hirn-Schranke assoziierte Krankheiten
- 3 Fachliteratur
- 4 Weblinks
- 5 Einzelnachweise
Unmittelbar mit der Blut-Hirn-Schranke assoziierte Erkrankungen
Während eine Vielzahl von neurologischen Erkrankungen die Blut-Hirn-Schranke stört oder schädigt, ist die Blut-Hirn-Schranke selbst nur bei einigen, äußerst seltenen, genetisch bedingten Syndromen der Ausgangspunkt einer Erkrankung.
GLUT1-Defizit-Syndrom
→ Hauptartikel: GLUT1-Defizit-Syndrom
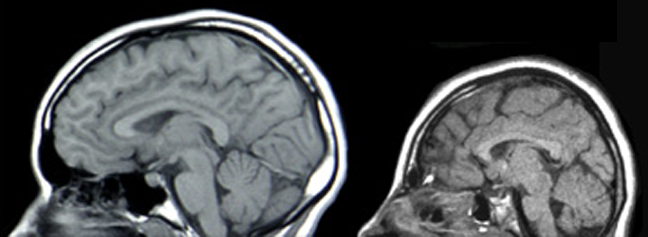
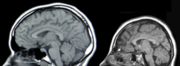 Kernspintomographische Aufnahme eines normalen Schädels (links) und einer Mikrozephalie (rechts). Die Mikrozephalie ist in diesem Fall durch eine Mutation des ASPM-Gens bedingt.
Kernspintomographische Aufnahme eines normalen Schädels (links) und einer Mikrozephalie (rechts). Die Mikrozephalie ist in diesem Fall durch eine Mutation des ASPM-Gens bedingt.Das GLUT1-Defizit-Syndrom ist eine äußerst seltene, autosomal-dominant[1] vererbte Krankheit. Die Krankheit manifestiert sich neonatal beziehungsweise im Kleinkindalter und wird durch einen Mangel an GLUT1-Transportern im Endothel der Blut-Hirn-Schranke hervorgerufen. Das Defizit an GLUT1-Transportern wird meist durch Neumutationen im SLC2A1-Gen verursacht. Dadurch wird das Gehirn nicht ausreichend mit D-Glucose versorgt und die betroffenen Patienten zeigen unter anderem eine Mikrozephalie, psychomotorische Retardierungen, Ataxie und eine Reihe anderer neurologischer Störungen.[2][3][4]
Biotin-ansprechende Basalganglienerkrankung
→ Hauptartikel: Biotin-ansprechende Basalganglienerkrankung
Wie beim GLUT1-Defizit-Syndrom ist auch bei der biotin-responsiven Basalganglienerkrankung eine Mutation in einem Gen, das für ein Transporter-Protein kodiert, die Ursache für die Erkrankung. Diese äußerst seltene Erkrankung – bisher sind nur etwas mehr als zehn Patienten mit dieser Erkrankung bekannt – wird durch einen Defekt im SLC19A3-Gen auf Chromosom 2, Genlocus q36.3 ausgelöst, das für einen Folat-Transporter kodiert.[5] Durch diesen Gendefekt wird bei den betroffenen Patienten das Gehirn nicht ausreichend mit Biotin versorgt, was sich durch eine subakute Enzephalopathie und vielfältige neurologische Symptome äußert.[1] Durch die Supplementation mit Biotin ist die Erkrankung gut therapierbar.[6]
Hereditäre Folat-Malabsorption
→ Hauptartikel: Hereditäre Folat-Malabsorption
Auch die autosomal-rezessiv[7] vererbte Folat-Malabsorption (lat. Malabsorption = „schlechte Aufnahme“) ist eine äußerst seltene Krankheit. Bei ihr wird das Gehirn mit Folsäure unterversorgt.[8] Ursache ist hier ein Defekt im PCFT-Gen (SLC46A1), das für einen protonen-abhängigen Folat-Transporter kodiert.[7][9]
MDR1-Defekt bei Collies und daraus abgeleiteten Hunderassen
→ Hauptartikel: MDR1-Defekt
Bei einigen Hunderassen, die sich alle vom Collie ableiten, besteht eine Überempfindlichkeit gegen manche Arzneimittel. Die Ursache dieser Überempfindlichkeit ist ein Defekt im mdr1-Gen, das für P-Glykoprotein kodiert. Durch diesen Gendefekt – mdr1-1Delta-Mutation genannt – ist der Efflux an der Blut-Hirn-Schranke weitgehend unterbunden und die entsprechenden Arzneimittel können mittels Diffusion in das Zentralnervensystem gelangen.[10] Die mdr1-1Delta-Mutation entstand vermutlich in der Mitte des 19. Jahrhunderts bei einem einzelnen Zuchthund und wurde von diesem an viele Generationen weitergegeben. Insbesondere bei der Gabe von Antiparasitika, Zytostatika und Antibiotika stellen sich bei den betroffenen Hunden starke neurotoxische Nebenwirkungen ein.[11]
Mittelbar mit der Blut-Hirn-Schranke assoziierte Krankheiten
Die Störung der Schutzwirkung der Blut-Hirn-Schranke ist eine Komplikation vieler neurodegenerativer Erkrankungen und Gehirnverletzungen. Auch einige Erkrankungen in der Peripherie, wie beispielsweise Diabetes mellitus oder Entzündungen, wirken sich schädlich auf die Funktion der Blut-Hirn-Schranke aus.[12]
Andere Erkrankungen wiederum stören die Funktion der Endothelien von „innen heraus“, das heißt die Integrität der Blut-Hirn-Schranke wird durch Einflüsse, die aus der extrazellulären Matrix heraus kommen, beeinträchtigt. Ein Beispiel hierfür ist das Glioblastom.[13]
Dagegen manifestiert sich eine Reihe von Erkrankungen im Gehirn dadurch, dass bestimmte Erreger die Blut-Hirn-Schranke überwinden können. Dazu gehören beispielsweise das HI-Virus, das Humane T-Zell-lymphotrope Virus Typ I, das West-Nil-Virus und Bakterien, wie Neisseria meningitidis oder Vibrio cholerae.[13]
Im Fall der Multiplen Sklerose sind die „Erreger“ Zellen der körpereigenen Immunabwehr, die die Blut-Hirn-Schranke überwinden. Ebenso überwinden bei einigen nicht-zerebralen Tumoren metastatisierende Zellen die Blut-Hirn-Schranke und können zu Metastasen im Gehirn führen.[13]
Diabetes mellitus
→ Hauptartikel: Diabetes mellitus
Der Diabetes mellitus (Zuckerkrankheit) ist eine Stoffwechselerkrankung, die eine Reihe von funktionellen und strukturellen Veränderungen verschiedener Organe und des Zentralnervensystems zur Folge hat. So finden auch an der Blut-Hirn-Schranke signifikante Veränderungen statt, die sowohl die Barriere-Wirkung, als auch die Transport-Funktionen beeinträchtigen. Die physikochemischen Eigenschaften der Plasmamembran und der Tight Junctions der Endothelien werden verändert.[14]
Mit zunehmender Dauer der Diabetes lässt die Barrierewirkung der Endothelien messbar nach. Dies wurde mit verschiedenen Markermolekülen unterschiedlicher Größe zunächst im Tiermodell festgestellt.[15] Dabei sind im wesentlichen die Strukturproteine der Tight Junctions betroffen.[16] Die Veränderungen der Tight Junctions sind nicht gleichmäßig über das Gehirn verteilt. Besonders betroffenen sind die Kapillaren des Mittelhirns, während andere Areale keine Veränderungen zeigen.[15] Die Auswirkungen dieser – durch Diabetes mellitus induzierten – strukturellen Veränderungen sind noch weitgehend unerforscht.[17] Allgemein bekannt sind die Auswirkungen der Diabetes auf periphere Endothelien, wo deren Funktionsstörungen die mit der Diabetes assoziierten Sekundärfolgen, wie Erblindung, chronisches Nierenversagen und Neuropathie, bewirken. Aktuelle klinische Studien zeigen für Diabetiker ein erhöhtes Risiko für eine vaskuläre Demenz, eine ventrikuläre Hypertrophie, einen lakunären Infarkt und eine Gehirnblutung, deren Ursachen in Veränderungen der Blut-Hirn-Schranke gesehen werden.[18] Offensichtlich sind Typ2-Diabetiker auch für die Entwicklung der Alzheimer-Erkrankung prädispositioniert.[19]
Multiple Sklerose
→ Hauptartikel: Multiple Sklerose
 VEGF löst die Bildung von vesikulovakuolären Organellen aus, über die eine transzelluläre Migration von Leukozyten durch das Endothel der Blut-Hirn-Schranke erfolgen kann. Die Darstellung zeigt ein Modell von VEGF-F.
VEGF löst die Bildung von vesikulovakuolären Organellen aus, über die eine transzelluläre Migration von Leukozyten durch das Endothel der Blut-Hirn-Schranke erfolgen kann. Die Darstellung zeigt ein Modell von VEGF-F.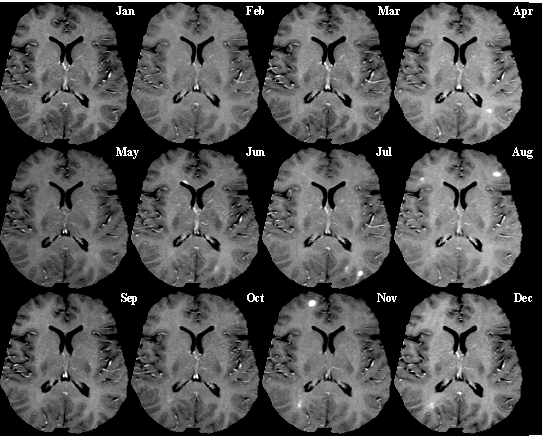 T1-gewichtete kontrastmittelverstärkte MRT bei einem Patienten mit Multipler Sklerose. Die Aufnahmen wurden in einem Abstand von einem Monat über ein Jahr aufgenommen. An frischen Entzündungsherden kann das Kontrastmittel die Blut-Hirn-Schranke passieren, wodurch die Entzündungsherde als weiße Flecken in den Aufnahmen sichtbar werden.
T1-gewichtete kontrastmittelverstärkte MRT bei einem Patienten mit Multipler Sklerose. Die Aufnahmen wurden in einem Abstand von einem Monat über ein Jahr aufgenommen. An frischen Entzündungsherden kann das Kontrastmittel die Blut-Hirn-Schranke passieren, wodurch die Entzündungsherde als weiße Flecken in den Aufnahmen sichtbar werden.Multiple Sklerose (MS) ist eine entzündliche demyelinisierende Autoimmunerkrankung des Zentralnervensystems (ZNS), bei der Lymphozyten und Makrophagen in das ZNS infiltriert werden. Bei der Infiltration überwinden diese körpereigenen Abwehrzellen die Blut-Hirn-Schranke. Die T-Lymphozyten greifen danach die im Interstitium des Gehirns oder Rückenmarks gelegenen Myelinscheiden an.
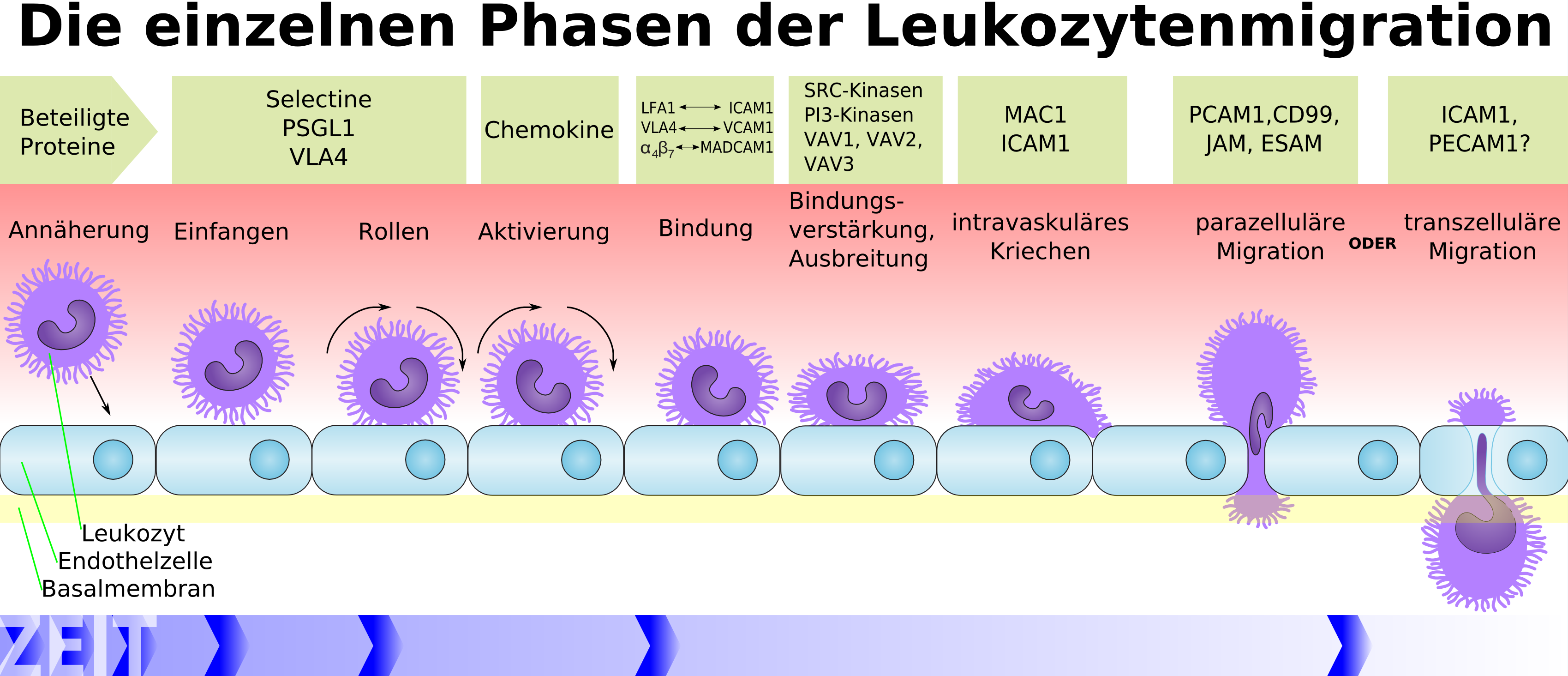
Die Einschleusung der Lymphozyten erfolgt über die Endothelzellen der Blut-Hirn-Schranke.[20] Der genaue Mechanismus dieses Vorgangs ist noch nicht vollständig geklärt. Durch die Entzündungsreaktion des Gewebes werden Zytokine ausgeschüttet, welche die Endothelzellen zur Expression der Adhäsionsmoleküle ICAM1 und VCAM1 anregen.[21][22] Die Zytokine bewirken bei den Endothelien zudem die Synthese von Chemokinen und Chemoattraktoren, die an der luminalen Seite der Kapillaren präsentiert werden. An der abluminalen Seite vorhandene Chemoattraktoren werden an die luminale Seite transportiert. Die an den Kapillarwänden entlang rollenden Leukozyten binden daraufhin an die Endothelien und es werden spezielle Adhäsionsmoleküle zur Verankerung der Leukozyten am Endothel exprimiert. Danach erfolgt entweder eine parazelluläre oder transzelluläre Transmigration.[23] Beide Wege sind möglich und beide Wege konnten nachgewiesen werden.[24][13]
Bei der parazellulären Transmigration – Diapedese genannt – öffnet der Leukozyt die Tight Junctions durch die Freisetzung von Signal- und Adhäsionsmolekülen. Die Endothelzellen ziehen sich dabei zusammen, die Tight Junctions öffnen sich und der Leukozyt kann so in das Interstitium des Zentralnervensystems eindringen. Der Vorgang selbst ist hochkomplex und hier nur stark vereinfacht wiedergegeben.[23] Auch der transzelluäre Weg ist möglich. Mehrere Vesikel und Vakuolen im Endothel formen dabei transendotheliale Zellkanäle. Diese vesikulovakuolären Organellen (VVO) finden sich in normalen Blutgefäßen, als auch in Tumorzellen.[25] Sie ermöglichen den aktiven Transport von Flüssigkeit, aber auch Zellen, über die Blut-Hirn-Schranke. Der Transport wird über den Vascular Endothelial Growth Factor (VEGF) vermittelt.[23] VEGF ist ein vasoaktives, permeabilitätsförderndes Protein. Der tranzelluläre Transport über die vesikulovakuolären Organellen ist offensichtlich auch ein Mechanismus mit dem Pathogene (Bakterien und Viren) die Blut-Hirn-Schranke überwinden können.[26][27]
Überhaupt ist die Fehlfunktion der Blut-Hirn-Schranke eines der Kennzeichen der Multiplen Sklerose. Vom Zusammenbruch der Blut-Hirn-Schranke sind vor allem junge Patienten mit einer besonders aggressiven Form der schubförmig remittierenden Multiplen Sklerose (RR-MS, engl. relapsing-remitting multiple sclerosis) betroffen. Das Phänomen lässt sich durch das Eindiffundieren eines paramagnetischen Kontrastmittels, zum Beispiel Gadopentetat-Dimeglumin (Gd-DTPA), bei der Magnetresonanztomographie sichtbar machen.[28] Das Verfahren ist dabei an Empfindlichkeit dem Monitoring von Markermolekülen wie Zytokinen oder Chemokinen überlegen.[29]
Die Ursache der Fehlfunktion der Blut-Hirn-Schranke bei der MS ist eine verminderte Expression der Tight-Junction-Proteine der Endothelzellen. Die Veränderungen an den Tight Junctions sind dabei nicht nur an den aktiven Läsionen zu beobachten, sondern auch – allerdings weniger häufig – in den inaktiven Läsionen und sogar in der Weißen Substanz. Die Veränderungen der Tight Junctions verschlechtern die Homöstase und haben offensichtlich einen Einfluss auf die Progression der Erkrankung, die Reparaturmechanismen und den Transport von Arzneimitteln.[30]
Ischämischer Schlaganfall
→ Hauptartikel: Ischämischer Schlaganfall
 Schematische Darstellung der Leukozytenmigration durch das Endothel.[23]
Schematische Darstellung der Leukozytenmigration durch das Endothel.[23]Beim ischämischen Schlaganfall (Hirninfarkt) kommt es infolge der zerebralen Ischämie (lokale Blutleere im Gehirn) und der anschließenden Reperfusion (Wiederherstellung des Blutzufuhr, siehe dazu auch Reperfusionsschaden) zu Veränderungen der Endothelien der Blut-Hirn-Schranke. Diese Veränderungen laufen in zwei Phasen ab. Die ersten Phase tritt wenige Minuten nach der Reperfusion ein, die zweite Phase mehrere Stunden nach der Ischämie.[31][32] Die Freisetzung von Oxidantien, proteolytischen Enzymen und Zytokinen ändert massiv die Durchlässigkeit der Blut-Hirn-Schranke und führt zur Bildung eines Ödems im Gehirn.[33] Aktivierte Leukozyten setzen als Folge des Ödems Matrix-Metalloproteasen (MMP) frei, die wiederum die Basallamina und Proteinkomplexe der Tight Junctions abbauen.[34] Hierbei spielt insbesondere MMP9 (Gelatinase B) eine wichtige Rolle.[35] Als Folge der Öffnung der Blut-Hirn-Schranke kann der neurotoxische gewebespezifische Plasminogenaktivator (t-PA) mittels passiver Diffusion in das Gehirn eindringen.[36][13]
Offensichtlich haben auch Leukozyten, die in das Parenchym des Gehirns migrieren, einen Anteil an den durch Ischämie und Reperfusion hervorgerufenen Schäden.[37][38] Die Leukozyten überwinden dabei die Endothelien – wie bei der Multiplen Sklerose – durch transendotheliale Migration.[39][40][13]
Bakterielle Infektionen des Zentralnervensystems (Meningitis)
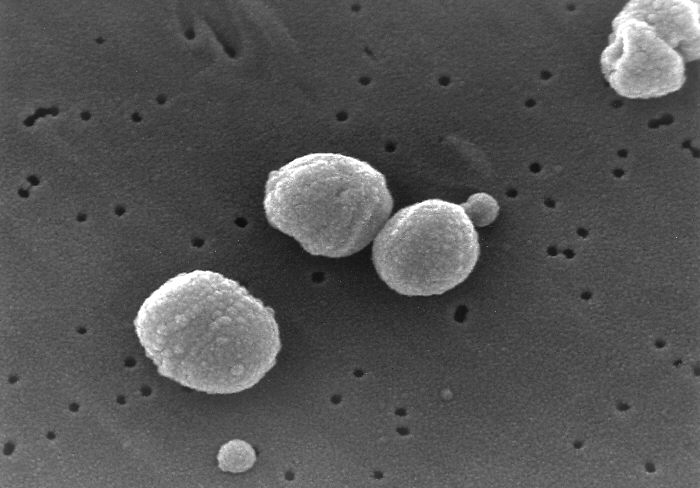
 Rasterelektronenmikrokopische Aufnahme von Streptococcus pneumoniae
Rasterelektronenmikrokopische Aufnahme von Streptococcus pneumoniae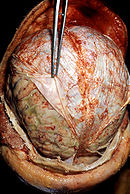 Durch Streptococcus pneumoniae verursachte eitrige Haubenmeningitis.
Durch Streptococcus pneumoniae verursachte eitrige Haubenmeningitis.Nur wenige durch das Blut übertragene Krankheitserreger sind prinzipiell in der Lage die Blut-Hirn-Schranke zu überwinden. Dazu gehören Meningokokken (Neisseria meningitidis), Streptokokken (Streptococcus agalactiae), Pneumokokken (Streptococcus pneumoniae), Haemophilus (Haemophilus influenzae), Listerien (Listeria monocytogenes) und Kolibakterien (Escherichia coli K1), die allesamt beim Überschreiten der Blut-Hirn-Schranke eine Hirnhautentzündung (Meningitis) auslösen können. Die genauen Mechanismen der Passage der Blut-Hirn-Schranke durch diese Pathogene sind noch nicht vollständig aufgeklärt. Die Pathogene nutzen verschiedene Wege zur Überwindung der Endothelien. Inflammatorische Prozesse spielen dabei eine wesentliche Rolle.[13]
Zunächst heften sich die Pathogene, wie beispielsweise L. monocytogenes, an den Endothelien fest und setzten dann eine Reihe von Lipopolysacchariden und Toxinen frei, was in den Endothelien wiederum die Produktion von verschiedenen Zytokinen (TNF-α, IL1β, PAF, TGFβ1), Matrix-Metalloproteasen und Caspasen anregt. Die Freisetzung dieser Proteine führt zu einer deutlichen Erhöhung der Permeabilität der Kapillaren.[13] Die erhöhte Permeabilität des Endothels ermöglicht die transendotheliale Migration von Leukozyten in das Gehirn. Diese setzen wiederum Zytokine und Matrix-Metalloproteasen frei, was eine weitere Aktivierung der Endothelien zur Folge hat und die Entzündungsreaktion in den betroffenen Bereichen des Zentralnervensystems erheblich verstärkt.[41][13]
S. pneumoniae ist dagegen in der Lage durch die Sezenierung von Pneumolysin, ein Enzym aus der Gruppe der Hämolysine, transmembrane Poren im Endothel zu erzeugen.[42] Über freigesetzte Endotoxine kann S. pneumoniae in den Endothelien sogar die Apoptose (programmierter Zelltod) auslösen.[43][44] Auch S. agalactiae attackiert direkt die Endothelien der Blut-Hirn-Schranke.[45][46]
N. meningitidis, S. pneumoniae und E. coli K1 sind darüber hinaus in der Lage mit Hilfe der transendothelialen Migration wie Leukozyten die Blut-Hirn-Schranke zu überwinden.[47][48] Nach ihrer Anheftung über Typ-IV-Pili an die Endothelzellen der Blut-Hirn-Schranke lösen sie eine ähnliche Signalkaskade wie Leukozyten aus, die letztlich die Migration dieser Pathogene in das Zentralnervensystem ermöglicht.[13]
Viren und die Blut-Hirn-Schranke
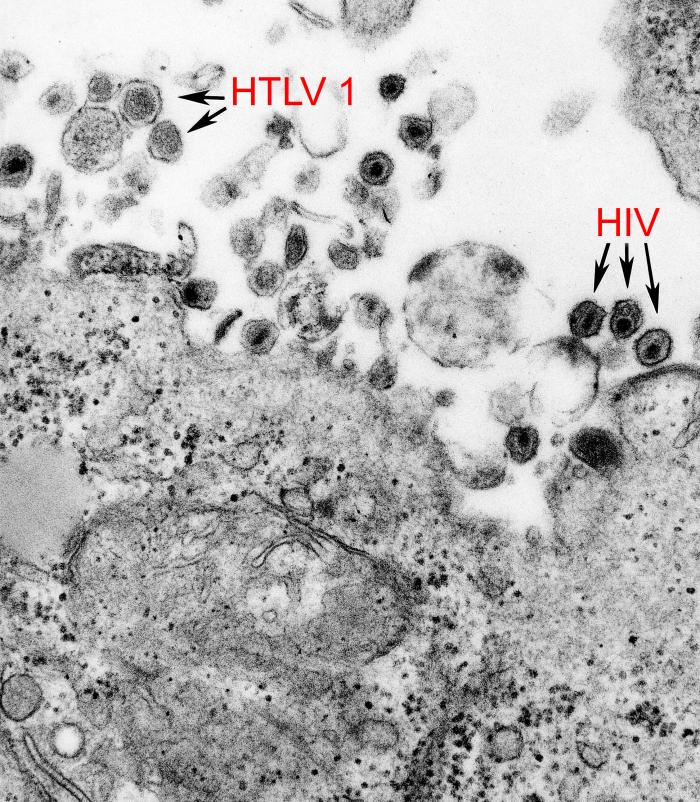
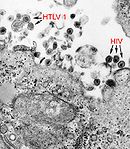 HTLV-1 und HIV-1 in einer transmissionselektronenmikroskopischen Aufnahme.
HTLV-1 und HIV-1 in einer transmissionselektronenmikroskopischen Aufnahme.Zu den Pathogenen, die in der Lage sind, die Blut-Hirn-Schranke zu überwinden, gehören auch Viren. Wie bei den Baktieren sind auch hier mehrere Mechanismen für die Passage in das Gehirn bekannt.[49]
Neurotrope Viren wie beispielsweise das Zytomegalievirus,[1] das humane Immundefizienz-Virus (HIV)[50] und das humane T-Zell-lymphotrope Virus Typ I (HTLV-I) können die Blut-Hirn-Schranke als freie Viren mittels Makropinozytose überwinden.[13]
Daneben spielt die transendotheliale Migration infizierter Leukozyten eine wesentliche Rolle für die Passage der Blut-Hirn-Schranke.[51][1] So wurde unter anderem im Fall des Zytomegalievirus[52] und des HI-Virus[53] dieser Mechanismus beschrieben. Zellen, die mit HIV oder HTLV-I infiziert sind, produzieren große Mengen an Matrix-Metalloproteasen, die wiederum die Basallamina angreifen und so die Migration der infizierten Zellen in das Gehirn mit ermöglichen.[54]
HIV
→ Hauptartikel: Humanes Immundefizienz-Virus
Das HI-Virus überwindet die Blut-Hirn-Schranke schon kurz nach der Infektion.[55] Offensichtlich spielen dabei mehrere, voneinander unabhängige, Mechanismen eine Rolle.[49] Die Migration über infizierte Monozyten und T-Lymphozyten („Trojanische Pferde“) ist im Fall des HI-Virus von entscheidender Bedeutung für die Infektion des Zentralnervensystems im späten Stadium von AIDS.[56][57] Der Mechanismus, mit dem das Virus mit Hilfe systemischer Leukozyten die Blut-Hirn-Schranke überwindet und nachhaltig schädigt ist noch unbekannt.[1] Das TAT-Protein, ein Genprodukt des HI-Virus, und gp120, ein vom HIV kodiertes Glykoprotein, spielen bei der nachfolgenden Schädigung des Zentralnervensystems eine wichtige Funktionen. Als Toxin ist TAT in der Lage direkt und indirekt oxidativen Stress[58] und inflammatorische Reaktionen im Endothel zu erzeugen.[59][60] Speziell der oxidative Stress ist ein wichtiger Faktor bei der Ausbildung der HIV-bedingten Demenz.[61] Darüber hinaus sind TAT und gp120 in der Lage in den Endothelzellen der Blut-Hirn-Schranke Apoptose auszulösen.[57]
Die Öffnung der Blut-Hirn-Schranke trägt dabei nicht nur zu einer Progression der Infektion mit HI-Viren im Gehirn bei, sondern führt auch zu Komplikationen im Krankheitsverlauf und zu Problemen bei der antiretroviralen Therapie.[55]
Humanes T-Zell-lymphotropes Virus Typ I
→ Hauptartikel: Humanes T-Zell-lymphotropes Virus Typ I
Auch das HTL-Virus I ist in der Lage die Blut-Hirn-Schranke zu passieren. Das Virus erhöht durch die Sekretion von Interleukin-1α und TNF-α sowohl die parazelluläre Permeabilität der Endothelien, als auch die Fähigkeit zur transzellulären Migration.[62] Die Genexpression der Tight-Junction-Proteine, wie beispielsweise ZO-1, wird dadurch ebenfalls beeinflusst. Offensichtlich sind die mit HTLV-1 infizierten Lymphozyten für die Öffnung der Blut-Hirn-Schranke und die damit verbundene weitere Infiltration von Lymphozyten, aber auch von neurotoxischen Plasmaproteinen, verantwortlich, die letztlich zur HTLV-I-assoziierten Myelopathie führen.[63][64]
West-Nil-Virus
→ Hauptartikel: West-Nil-Virus
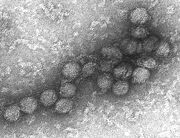 Das West-Nil-Virus im Transmissions-Elektronenmikroskop.
Das West-Nil-Virus im Transmissions-Elektronenmikroskop.Das West-Nil-Virus ist ein behülltes RNA-Virus, das ebenfalls in der Lage ist die Blut-Hirn-Schranke zu überwinden und eine Enzephalitis oder Meningitis auslösen zu können. Das Virus infiziert dabei im peripheren lymphatischen Gewebe Makrophagen oder dendritische Zellen. Diese geben daraufhin TLR3-abhängige (Toll-like Receptor) antivirale und immunmodulierende Zytokine wie Interferone, Interleukin-6 und TNF-α ab, wodurch eine weitere Infektion im peripheren Gewebe unterbunden wird. Die TLR3-abhängige Freisetzung von TNF-α ermöglicht dem West-Nil-Virus die Überwindung der Blut-Hirn-Schranke. Dabei spielen durch TNF-α ausgelöste Veränderungen der Tight Junctions eine wichtige Rolle. Durch diese Veränderungen können entweder die freien Viren oder aber virusinfizierte Leukozyten in das Zentralnervensystem eindringen.[65][66]
Neurodegenerative Erkrankungen
Bis zu Beginn des 21. Jahrhunderts ist man davon ausgegangen, dass neurodegenerative Erkrankungen, wie die Alzheimer- und Parkinson-Krankheit, keinen Einfluss auf die Blut-Hirn-Schranke haben. Neuere Untersuchungsergebnisse widerlegen diese These. Mit Hilfe der kontrastmittelverstärkten Magnetresonanztomographie und biochemischer Untersuchungen des Liquors konnten beispielsweise bei Alzheimer-Patienten, im Vergleich zu einer Gruppe gleichaltriger nicht erkrankter Personen, funktionale Veränderungen der Blut-Hirn-Schranke in Richtung einer erhöhten Durchlässigkeit festgestellt werden.[67] Die erhöhte Permeabilität ist offensichtlich schon bei einem sehr frühen Stadium der Erkrankung vorhanden.[68] Die Konsequenzen dieser Erkenntnisse werden derzeit noch kontrovers diskutiert.[69] Die Veränderungen an der Blut-Hirn-Schranke könnten Einfluss auf die Progression der Erkrankung, sowie mögliche zukünftige Therapieansätze haben.[70]
Glioblastom und andere primäre Hirntumoren
→ Hauptartikel: Glioblastom
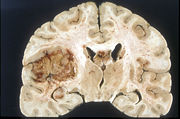 Koronarer Schnitt durch ein Gehirn mit einem Glioblastom (linke Hälfte, grau-rot)
Koronarer Schnitt durch ein Gehirn mit einem Glioblastom (linke Hälfte, grau-rot)Das Wachstum eines Tumors ist schon in einer sehr frühen Phase von der Neubildung von Blutgefäßen (Neovaskularisation) begleitet, damit der Tumor ausreichend mit Sauerstoff und Nährstoffen versorgt werden kann. Bereits ab einem Durchmesser von 1 bis 2 mm beginnt die Neovaskularisation.[71] Die neugebildeten Blutgefäße weisen erhebliche strukturelle Unterschiede gegenüber den normalen Blutgefäßen auf. Bei Hirntumoren führen diese strukturellen Unterschiede zu signifikanten lokalen Veränderungen der Blut-Hirn-Schranke. Speziell beim Glioblastom ist die Neovaskularisation sehr stark ausgeprägt und mit ein Faktor für das aggressive Wachstum bei dieser Krebserkrankung.[72][13]
Die neugebildeten Blutgefäße primärer Hirntumoren weisen eine gewundenere Struktur als die normalen Blutgefäße des Gehirns auf. Die Endothelien sind von einer verformten Basallamina überzogen und exprimieren nicht mehr die Tight-Junction-Proteine Claudin-3 und Occludin.[73][74] Dem gegenüber wird in den Tumoren in großen Mengen der Vascular Endothelial Growth Factor (VEGF) produziert[75][76], der die Endozytose des Zelladhäsionsproteins VE-Cadherin fördert und dadurch die Durchlässigkeit der Endothelien weiter erhöht.[77][13] Der Expressionsgrad von Occludin korreliert umgekehrt proportional mit dem Grading und der Durchlässigkeit der betroffenen Endothelien für Kontrastmittel, die eine gesunde Blut-Hirn-Schranke nicht passieren können.[74] Die erhöhte Permeabilität der Endothelzellen für bestimmte Kontrastmittel wird in der Diagnostik angewendet (siehe dazu den Absatz Humandiagnostik).
In der Therapie sind die Blutgefäße der Hirntumoren ein potenzielles Target für Angiogenese-Inhibitoren.[78][79][72] Die Zielstrukturen sind dabei unter anderem die αvβ3-Integrine (Cilengitid) und VEGF (Bevacizumab).
Metastatisierende Tumorzellen
→ Hauptartikel: Hirnmetastase
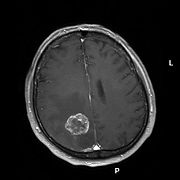 T1-gewichtete kontrastmittelverstärkte Magnetresonanztomografie einer Hirnmetatase eines Bronchialkarzinoms (linke Hirnhälfte, weißer kreisrunder Bereich
T1-gewichtete kontrastmittelverstärkte Magnetresonanztomografie einer Hirnmetatase eines Bronchialkarzinoms (linke Hirnhälfte, weißer kreisrunder BereichMetastatisierende Tumorzellen, speziell von Bronchialkarzinomen, Mammakarzinomen und malignen Melanomen, sind in der Lage die Blut-Hirn-Schranke zu überwinden und zerebrale Metastasen mit ausnahmslos schlechter Prognose zu bilden.[13] Bei etwa 20 bis 40 % aller Krebspatienten bilden sich Hirnmetastasen.[80] Bei der Migration dieser Zellen in das Zentralnervensystem spielt ebenfalls der Vascular Endothelial Growth Factor (VEGF) eine wichtige Rolle. VEGF wird von Mammakarzinom-Zellen[81] und Melanom-Zellen[82] besonders stark exprimiert.[13]
Einige metastatisierende Tumorzellen exprimieren – ähnlich den aktivierten Leukozyten – ein ganzes Sortiment von Adhäsionsmolekülen an ihrer Zelloberfläche. Zusammen mit Chemokinrezeptoren, wie beispielsweise CXCR4, das an das im Gehirn stark exprimierte Chemokin CXCL12 anbindet, gibt dies den metastatisierten Tumorzellen die Möglichkeit sich an das Endothel der Kapillargefäße anzuheften.[83] Nach der Anbindung werden die Enothelien von den Tumorzellen aktiviert, wodurch die Migration in das Zentralnervensystem ermöglicht wird. In vitro konnte die Funktion von CXCR4 durch Blockade des intrazellulären Wegs mittels Phosphoinositid-3-Kinasen nachgewiesen werden. Tumorzellen der Brustkrebszellinie DU4475 mit blockiertem CXCR4 konnten nicht mehr durch kultivierte Endothelien migrieren.[84]
Fachliteratur
- D. Kobiler u. a.: Blood-brain Barrier. Verlag Springer, 2001, ISBN 0-306-46708-9
- W. M. Pardridge: Introduction to the Blood-brain Barrier. Cambridge University Press, 1998, ISBN 0-521-58124-9
Weblinks
Einzelnachweise
- ↑ a b c d e J. Klepper u. a.: Autosomal dominant transmission of GLUT1 deficiency. In: Hum Molec Genet 10, 2001, S. 63–68. PMID 11136715
- ↑ D. C. De Vivo u. a.: Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. In: NEJM 325, 1991, S. 703–709. PMID 1714544
- ↑ oprha.net: Enzephalopathie durch GLUT1-Defekt. eingesehen am 3. März 2009
- ↑ Online Mendelian Inheritance in Man: Glucose Transport Defect, Blood-Brain Barrier. eingesehen am 3. März 2009
- ↑ W. Q. Zeng u. a.: Biotin-responsive basal ganglia disease maps to 2q36.3 and is due to mutations in SLC19A3. In: Am J Hum Genet 77, 2005, S. 16–26. PMID 15871139
- ↑ Online Mendelian Inheritance in Man: Basal Ganglia Disease, Biotin-responsive. eingesehen am 3. März 2009
- ↑ a b R. Zhao u. a.: The spectrum of mutations in the PCFT gene, coding for an intestinal folate transporter, that are the basis for hereditary folate malabsorption. In: Blood 110, 2007, S. 1147–1152. PMID 17446347
- ↑ P. C. Su: Congenital folate deficiency. In: NEJM 294, 1976, S. 1128. PMID 176588
- ↑ Online Mendelian Inheritance in Man: Folic Acid, Transport Defect Involving. eingesehen am 3. März 2009
- ↑ S. Fecht und O. Distl: Review of prevalence, genetic aspects and adverse effects of the mdr1-1Delta mutation in dogs. In: Dtsch Tierarztl Wochenschr 115, 2008, S. 212–219. PMID18605373 (Review)
- ↑ J. Geyer u. a.: MDR1-Defekt. Multiple Medikamentenüberempfindlichkeit bei Britischen Hütehunden. In: Kleintier konkret 9, 2006, S. 16–20.
- ↑ B. T. Hawkins und R. D. Egleton: Pathophysiology of the blood-brain barrier: animal models and methods. In: Curr Top Dev Biol 80, 2008, S. 277–309. PMID 17950377 (Review)
- ↑ a b c d e f g h i j k l m n o N. Weiss u.a.: The blood-brain barrier in brain homeostasis and neurological diseases. In: Biochim Biophys Acta 2008, [Epub ahead print] PMID 19061857 (Review)
- ↑ M. H. Horani und A. D. Mooradian: Effect of diabetes on the blood brain barrier. In: Curr Pharm Des 9, 2003, S. 833–840. PMID 12678883 (Review)
- ↑ a b J. D. Huber u. a.: Streptozotocin-induced diabetes progressively increases blood-brain barrier permeability in specific brain regions in rats. In: Am J Physiol Heart Circ Physiol 291, 2006, S. H2660–H2668. PMID 16951046
- ↑ J. M. Chehade u. a.: Diabetes-related changes in rat cerebral occlusion and zonula occludens-1 (ZO-1) expression. In: Neurochem Res 27, 2002, S. 249–252. PMID 11958524
- ↑ W. A. Banks: The dam breaks: disruption of the blood-brain barrier in diabetes mellitus. In: Am J Physiol Heart Circ Physiol 291, 2006, S. 2595–2596. PMID 16877556 (Review)
- ↑ J. D. Huber: Diabetes, cognitive function, and the blood-brain barrier. In: Curr Pharm Des 14, 2008, S. 1594–1600. PMID 18673200 (Review)
- ↑ P. A. Whitmer: Type 2 diabetes and risk of cognitive impairment and dementia. In: Curr Neurol Neurosci Rep 7, 2007, S. 373–380. PMID 17764626 (Review)
- ↑ J. Correale und A. Villa: The blood-brain-barrier in multiple sclerosis: functional roles and therapeutic targeting. In: Autoimmunity 40, 2007, S. 148–160. PMID 17453713
- ↑ J. J. Campbell u. a.: Biology of chemokine and classical chemoattractant receptors. Differential requirements for adhesion-triggering versus chemotactic responses in lymphoid cells. In: J Cell Biol 134, 1996, S. 255–266. PMID 8698820
- ↑ J. J. Campbell u. a.: Chemokines and the arrest of lymphocytes rolling under flow conditions. In: Science 279, 1998, S. 381–384. PMID 9430588
- ↑ a b c d K. Ley u. a.: Getting to the site of inflammation: the leukocyte adhesion cascade updated. In: Nature Reviews Immunology 7, 2007, S. 678–689. PMID 17717539 (Review)
- ↑ C. V. Carman und T. A. Springer: Trans-cellular migration: cell–cell contacts get intimate. In: Curr Opin Cell Biol 20, 2008, S. 533–540. PMID 18595683 (Review)
- ↑ A. M. Dvorak u. a.: The vesiculo-vacuolar organelle (VVO): A distinct endothelial cell structure that provides a transcellular pathway for macromolecular extravasation. In: J Leukoc Biol 59, 1996, S. 100–115. PMID 8558058
- ↑ A. S. Lossinsky und R. R. Shivers: Structural pathways for macromolecular and cellular transport across the blood-brain barrier during inflammatory conditions. In: Histol Histopathol 19, 2004, S. 535–564. PMID 15024715 (Review)
- ↑ S. Nag: The blood-brain barrier. Humana Press, 2003, S. 76. ISBN 1-58829-073-5
- ↑ L. A. Stone: Blood-brain barrier disruption on contrast-enhanced MRI in patients with mild relapsing-remitting multiple sclerosis: relationship to course, gender and age. In: Neurology 45, 1995, S. 1122–1126. PMID 7783875
- ↑ E. Waubant: Biomarkers indicative of blood-brain barrier disruption in multiple sclerosis. In: Dis Markers 22, 2006, S. 235–244. PMID 17124345 (Review)
- ↑ S. McQuaid u. a.: The effects of blood-brain barrier disruption on glial cell function in multiple sclerosis. In: Biochem Soc Trans 37, 2009, S. 329–331. PMID 19143657 (Review)
- ↑ L. Belayev u. a.: Quantitative evaluation of blood-brain barrier permeability following middle cerebral artery occlusion in rats. In: Brain Res 739, 1996, S. 88–96. PMID 8955928
- ↑ J. Aronowski u. a.: Reperfusion injury: demonstration of brain damage produced by reperfusion after transient focal ischemia in rats. In: J Cereb Blood Flow Metab 17, 1997, S. 1048–1056. PMID 9346429
- ↑ U. Dirnagl u. a.: Pathobiology of ischaemic stroke: an integrated view. In: Trends Neurosci 22, 1999, S. 391–397. PMID 10441299 (Review)
- ↑ V. W. Yong: Metalloproteinases: mediators of pathology and regeneration in the CNS. In: Nat Rev Neurosci 6, 2005, S. 931–944. PMID 16288297 (Review)
- ↑ M. Asahi u. a.: Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. In: J Neurosci 21, 2001, S. 7724–7732. PMID 11567062
- ↑ K. Benchenane u. a.: Tissue-type plasminogen activator crosses the intact blood-brain barrier by low-density lipoprotein receptor-related protein-mediated transcytosis. In: Circulation 111, 2005, S. 2241–2249. PMID 15851587
- ↑ S. Kuroda und B. K. Siesjö: Reperfusion damage following focal ischemia: pathophysiology and therapeutic windows. In: Clin Neurosci 4, 1997, S. 199–212. PMID 9186042 (Review)
- ↑ A. M. Planas u. a.: Signalling pathways mediating inflammatory responses in brain ischaemia. In: Biochem Soc Trans 34, 2006, S. 1267–1270. PMID 17073799 (Review)
- ↑ P. J. Lindsberg u. a.: Endothelial ICAM-1 expression associated with inflammatory cell response in human ischemic stroke. In: Circulation 94, 1996, S. 939–945. PMID 8790029
- ↑ R. L. Zhang u. a.: The temporal profiles of ICAM-1 protein and mRNA expression after transient MCA occlusion in the rat. In: Brain Res 682, 1995, S. 182–188. PMID 7552309
- ↑ S. L. Leib u. a.: Matrix metalloproteinases contribute to brain damage in experimental pneumococcal meningitis, Infect. In: Immun 68, 2000, S. 615–620. PMID 10639424
- ↑ G. Zysk u. a.: Pneumolysin is the main inducer of cytotoxicity to brain microvascular endothelial cells caused by Streptococcus pneumoniae. In: Infect Immun 69, 2001, S. 845–852. PMID 11159977
- ↑ A. Halle: Streptococcus pneumoniae induziert Apoptose in zerebralen Endothelzellen: Die Rolle bakterieller Toxine. Dissertation, Medizinische Fakultät der Charité, 2005.
- ↑ D. D. Bannerman und S. E. Goldblum: Direct effects of endotoxin on the endothelium: barrier function and injury. In: Lab Invest 79, 1999, S. 1181–1199. PMID 10532583 (Review)
- ↑ K. S. Doran u. a.: Blood–brain barrier invasion by group B streptococcus depends upon proper cell-surface anchoring of lipoteichoic acid. In: J Clin Invest 115, 2005, S. 2499–2507 PMID 16138192
- ↑ H. C. Maisey u. a.: Group B streptococcal pilus proteins contribute to adherence to and invasion of brain microvascular endothelial cells. In: J Bacteriol 189, 2007, S. 1464-1467. PMID 17041051
- ↑ K. S. Kim: Pathogenesis of bacterial meningitis: from bacteraemia to neuronal injury. In: Nat Rev Neurosci 4, 2003, S. 376–385. PMID 12728265 (Review)
- ↑ X. Nassif u. a.: How do extracellular pathogens cross the blood-brain barrier? In: Trends Microbiol 10, 2002, S. 227–232. PMID 11973156 (Review)
- ↑ a b D. T. Wu u. a.: Mechanisms of leukocyte trafficking into the CNS. In: J Neurovirol 6, 2000, S. 82-85. PMID 10871769
- ↑ W. A. Banks u. a.: Transport of human immunodeficiency virus type 1 pseudoviruses across the blood-brain barrier: role of envelope proteins and adsorptive endocytosis. In: J Virol 75, 2001, S. 4681–4691. PMID 11312339
- ↑ A. T. Haase: Pathogenesis of lentivirus infections. In: Nature 322, 1986, S. 130–136. PMID 2425264 (Review)
- ↑ J. D. Reuter u. a.: CD4+ T-cell reconstitution reduces cytomegalovirus in the immunocompromised brain. In: J Virol 79, 2005, S. 9527-9539. PMID 16014915
- ↑ A. Alexaki und B. Wigdahl: HIV-1 infection of bone marrow hematopoietic progenitor cells and their role in trafficking and viral dissemination. In: PLoS Pathog 4, 2008, S. e1000215. PMID 19112504
- ↑ K. Conant u. a.: Cerebrospinal fluid levels of MMP-2, 7, and 9 are elevated in association with human immunodeficiency virus dementia. In: Ann Neurol 46, 1999, S. 391–398. PMID 10482270
- ↑ a b J. R. Berger und M. Avison: The blood brain barrier in HIV infection. In: Front Biosci 9, 2004, S. 2680-2685. PMID 15358591
- ↑ H. S. Nottet u. a.: Mechanisms for the transendothelial migration of HIV-1-infected monocytes into brain. In: J Immunol 156, 1996, S. 1284-1295. PMID 8558009
- ↑ a b T. A. Kim u. a.: HIV-1 Tat-mediated apoptosis in human brain microvascular endothelial cells. In: J Immunol 170, 2003, S. 2629-2637. PMID 12594291
- ↑ C. B. Pocernich u. a.: HIV-dementia, Tat-induced oxidative stress, and antioxidant therapeutic considerations. In: Brain Res Brain Res Rev 50, 2005, S. 14-26. PMID 15890409 (Review)
- ↑ M. Toborek u. a.: HIV-Tat protein induces oxidative and inflammatory pathways in brain endothelium. In: J Neurochem 84, 2003, S. 169-179. PMID 12485413
- ↑ F. M. Hofman u. a.: Exogenous Tat protein activates human endothelial cells. Blood 92, 1993, S. 2774-2780. PMID 7693046
- ↑ J. Steiner u. a.: Oxidative stress and therapeutic approaches in HIV dementia. In: Antioxid Redox Signal 8, 2006, S. 2089-2100. PMID 17034352 (Review)
- ↑ P. V. Afonso u. a.: Alteration of blood-brain barrier integrity by retroviral infection. In: PLoS Pathog 4, 2008, e1000205. PMID 19008946
- ↑ P. V. Afonso u. a.: Human blood-brain barrier disruption by retroviral-infected lymphocytes: role of myosin light chain kinase in endothelial tight-junction disorganization. In: J Immunol 179, 2007, S. 2576–2583. PMID 17675520
- ↑ P. Shapshak u. a.: HTLV-III Can Cross the Blood-Brain Barrier. In: Annals of the New York Academy of Sciences 529, 2006, S. 291–294. doi:10.1111/j.1749-6632.1988.tb51485.x
- ↑ M. S. Diamond und R. S. Klein: West Nile virus: crossing the blood-brain barrier. In: Nature Medicine 10, 2004, S. 1294–1295. PMID 15580248
- ↑ R. Paterson: How West Nile virus crosses the blood-brain barrier. In: Lancet Neurol 4, 2005, S. 18. PMID 15645594
- ↑ A. J. Farrall und J. M. Wardlaw: Blood-brain barrier: Ageing and microvascular disease — systematic review and meta-analysis. In: Neurobiol Aging 30, 2009, S. 337-352. PMID 17869382
- ↑ J. M. Starr u. a.: Blood-brain barrier permeability in Alzheimer's disease: a case-control MRI study. In: Psychiatry Res 171, 2009, S. 232-41. PMID 19211227
- ↑ B. V. Zlokovic: Neurovascular mechanisms of Alzheimer's neurodegeneration. In: Trends Neurosci 28, 2005, S. 202–208. PMID 15808355 (Review)
- ↑ B.S. Desai u. a.: Blood-brain barrier pathology in Alzheimer's and Parkinson's disease: implications for drug therapy. In: Cell Transplant 16, 2007, S. 285–299. PMID 17503739
- ↑ D. Hanahan und J. Folkman: Patterns and emering mechanisms of the angiogenic switch during tumorigenesis. In: Cell 86, 1996, S. 353–364. PMID 8756718 (Review)
- ↑ a b J. C. Anderson u. a.: New molecular targets in angiogenic vessels of glioblastoma tumours. In: Expert Rev Mol Med 10, 2008, S. e23. PMID 1868433 (Review)
- ↑ H. Wolburg u. a.: Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. In: Acta Neuropathol 105, 2003, S. 586–592. PMID 12734665
- ↑ a b M. C. Papadopoulos u. a.: Occludin expression in microvessels of neoplastic and non-neoplastic human brain. In: Neuropathol Appl Neurobiol 27, 2001, S. 384–395. PMID 11679090
- ↑ R. K. Jain u. a.: Angiogenesis in brain tumours. In: Nat Rev Neurosci 8, 2007, S. 610–622. PMID 17643088 (Review)
- ↑ K. H. Plate u. a.: Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. In: Nature 359, 1992, S. 845–848. PMID 1279432
- ↑ J. Gavard und J.S. Gutkind: VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. In: Nat Cell Biol 8, 2006 1223–1234. PMID 17060906
- ↑ T. Würdinger und B. A. Tannous: Glioma angiogenesis: Towards novel RNA therapeutics. In: Cell Adh Migr 3, 2009, [Epub ahead of print] PMID 19262177
- ↑ T. Visted u. a.: Mechanisms of tumor cell invasion and angiogenesis in the central nervous system. In: Front Biosci 8, 2003, S. e289–304. PMID 12700036 (Review)
- ↑ M. Prados und C. Wilson: Neoplasms of the central nervous system. In: Cancer Medicine Verlag Lea&Febiger, S. 1080-1119, 1993. ISBN 0-8121-1422-1.
- ↑ T. H. Lee u. a.: Vascular endothelial growth factor modulates the transendothelial migration of MDA-MB-231 breast cancer cells through regulation of brain microvascular endothelial cell permeability. In: J Biol Chem 278, 2003, S. 5277–5284. PMID 12446667
- ↑ D. Marchetti u. a.: Brain-metastatic melanoma: a neurotrophic perspective. In: Pathol Oncol Res 9, 2003, S. 147–158. PMID: 14530807 (Review)
- ↑ B. C. Lee u. a.: Involvement of the chemokine receptor CXCR4 and its ligand stromal cell-derived factor 1alpha in breast cancer cell migration through human brain microvascular endothelial cells. In: Mol Cancer Res 2, 2004, S. 327–338. PMID 15235108
- ↑ N. Vykhodtseva u. a.: Progress and problems in the application of focused ultrasound for blood-brain barrier disruption. In: Ultrasonics 48, 2008, S. 279–296. PMID 18511095





Wikimedia Foundation.