- Akute lymphoblastische Leukämie
-
Klassifikation nach ICD-10 C91.0 Akute lymphoblastische Leukämie
ICD-O 9835/3 (B-Linie)
ICD-O 9837/3 (T-Linie)ICD-10 online (WHO-Version 2006) Die akute lymphatische Leukämie (syn. akute lymphoblastische Leukämie, kurz ALL) ist eine akute Leukämie, die von bösartig entarteten Vorläuferzellen der Lymphozyten ausgeht. Verschiedene Symptome wie allgemeine Schwäche mit Blutarmut, Blutgerinnungsstörungen, Fieber mit schwer verlaufenden Infekten, Knochenschmerzen u. a. m. können auftreten. Die Behandlung erfolgt mittels Chemotherapie. Führte die ALL noch vor 30 Jahren bei der ganz überwiegenden Zahl der Patienten innerhalb von wenigen Wochen zum Tode, so ist sie heute bei 40–50 % der Erwachsenen und bei ca. 70-80 % aller Kinder mit intensiver Chemotherapie heilbar.

Inhaltsverzeichnis
Häufigkeit
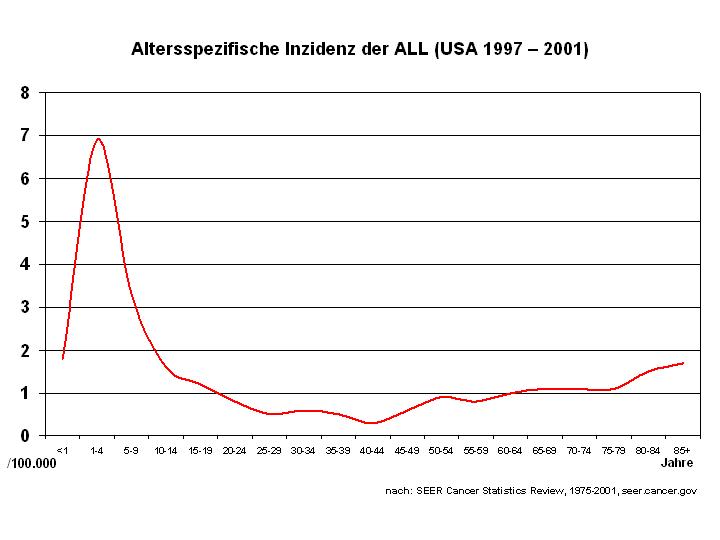
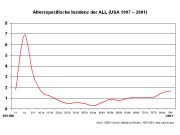 Altersspezifische Inzidenz der ALL[1]
Altersspezifische Inzidenz der ALL[1]Die ALL ist eine seltene Erkrankung mit einer Inzidenz bezogen auf alle Altersgruppen von etwa 1,5 Neuerkrankungen/100.000 im Jahr. Es besteht ein Übergewicht von männlichen Erkrankten (1,4 zu 1). Für Deutschland schätzt man etwa 500 Neuerkrankungen bei Erwachsenen und ebenfalls ca. 500 Neuerkrankungen bei Kindern pro Jahr. Genaue Zahlen existieren für Erwachsene wegen des Fehlens eines zentralen Krebsregisters nicht, die Zahlen für Kinder unter 15 Jahren beruhen auf den Daten des Deutschen Kinderkrebsregisters, in dem schätzungsweise 90 % aller Fälle erfasst sind.[2] In der Schweiz und in Österreich kann man jeweils etwa 40–50 Neuerkrankungen bei Erwachsenen und Kindern pro Jahr annehmen.[3] Für die Vereinigten Staaten werden die Neuerkrankungen im Jahr 2007 auf ungefähr 5.200 Fälle geschätzt.[4] Das lebenslange Risiko (lifetime risk), an akuter lymphatischer Leukämie zu erkranken, liegt damit bei ungefähr 1 zu 838, d. h. etwa eine Person unter 838 wird im Laufe ihres Lebens an ALL erkranken. Die Verteilung über die Altersgruppen zeigt einen Gipfel im Kindesalter (6,5/100.000 bei Kindern unter 4 Jahren) und einen zweiten, geringeren im höheren Alter (1,5/100.000 bei über 80-Jährigen). Bei Erwachsenen macht die ALL weniger als 15 % aller akuten Leukämien aus (d. h. mehr als 85 % aller Erkrankung sind akute myeloische Leukämien). Bei Kindern ist dies umgekehrt und die ALL ist damit die häufigste maligne Erkrankung im Kindesalter.
Ursachen und Entstehung
Ursache der Erkrankung sind genetische Veränderungen in einer lymphatischen Zelle, die zur malignen (bösartigen) Transformation dieser Zelle führen. Diese genetischen Veränderungen sind (von seltenen Spezialfällen abgesehen) im Laufe des Lebens erworben und weder vererbt noch vererbbar, da die Keimbahnzellen (Eizellen, Spermien) nicht betroffen sind. Die genaue Kausalkette, die zum Auftreten der Erkrankung führt, ist bisher nicht bekannt. Es gibt Risikofaktoren für das Entstehen von Leukämien (ionisierende Strahlung, chemische Mutagene etc.), aber bei den allermeisten Patienten kann man trotz sorgfältiger Suche keine spezifische Ursache finden. Auch gibt es bisher keinen Beweis für eine mögliche infektiöse Ursache, z. B. durch Viren. Die einzige Ausnahme bildet die adulte T-Zell-Leukämie in Japan, die durch das Retrovirus HTLV-I verursacht wird, das aber in Europa so gut wie gar nicht vorkommt. Die maligne transformierte Zelle und ihre durch Zellteilung entstandenen Tochterzellen vermehren sich unkontrolliert und ungebremst und verdrängen die normale Blutbildung (Hämatopoese) im Knochenmark. Die ALL ist also eine klonale Erkrankung, d. h. alle ALL-Zellen sind nahezu identische genetische Kopien voneinander. Die malignen Zellen sammeln sich vorwiegend in den lymphatischen Organen (v. a. Lymphknoten, Milz, Thymus), jedoch auch anderen Organen wie z. B. dem Zentralnervensystem (ZNS), an. Es kommt zu einer zunehmenden Störung der Blutbildung (Knochenmarkinsuffizienz) mit Blutarmut (Anämie) und Thrombozytopenie (Fehlen von Thrombozyten) und einer damit verbundenen Blutungsneigung, sowie zu einer Immunschwäche. Unbehandelt verläuft die Erkrankung schnell tödlich.
Symptome
Die Symptome bei der ALL ähneln denen der akuten myeloischen Leukämie.
Es treten auf:
- Störungen der Blutbildung:
- Blutarmut (Anämie) mit allgemeiner Schwäche, Leistungsknick, Abgeschlagenheit
- Mangel an Thrombozyten (Blutplättchen), u. U. verbunden mit Blutungsneigung, z. B. Spontanblutungen jeder Art
- starke Abwehrschwäche durch relativen Mangel an funktionsfähigen weißen Blutzellen (Leukozyten) mit schwer verlaufenden Infektionen
- häufig Hepatosplenomegalie (Vergrößerung von Leber und Milz)
- häufig Schwellungen der Lymphknoten (allerdings nicht so ausgeprägt wie bei der chronischen lymphatischen Leukämie)
- manchmal Knochenschmerzen, die auch Erstsymptom der Erkrankung sein können (insbesondere bei Kindern)
- in ca. 10 % der Fälle Meningeosis leucaemica (Befall des Zentralnervensystems durch Leukämiezellen) u. U. mit neurologischen Ausfällen
- Thymusschwellung (sogenannter Mediastinaltumor) häufig bei der T-ALL mit u. U. oberer Einflussstauung
Zum Diagnosezeitpunkt zeigen Kinder mit einer ALL folgende relative Häufigkeiten klinischer Zeichen, Symptome und typischer Laborbefunde. Letztere spiegeln dabei das Ausmaß der Störung der Blutbildung durch die ALL wieder, da die normale Blutbildung im Knochenmark durch die ALL verdrängt wird (sogenannte „Verdrängungsmyelopathie“).
Tabelle 1. Klinische Zeichen und Symptome bei Kindern mit akuter lymphatischer Leukämie[5] Symptome Prozent der Patienten Fieber (Temperatur >38,5 °C) 61 % Blutungszeichen (z. B. Petechien oder Purpura) 48 % Knochenschmerzen 23 % Lymphknotenschwellungen (Lymphadenopathie) 50 % Alleinige Milzvergrößerung (Splenomegalie) 63 % Milz- und Lebervergrößerung (Hepatosplenomegalie) 68 % Tabelle 2. Laboruntersuchungsbefunde bei Kindern mit akuter lymphatischer Leukämie[6] Laboruntersuchungsbefunde Wertebereich Prozent der Patienten Leukozyten; WBC (white blood cells), „Leukos“ Normal: 4.000/µL-12.000/µL * <10.000/µL 53 % 10.000 bis 49.000/µL 30 % >50.000/µL 17 % Hämoglobin; HGB, Hb Normal: 11,0–16,0 g/dL * <7,0 g/dL 43 % 7,0 bis 11,0 g/dL 45 % >11,0 g/dL 12 % Thrombozyten; PLT (platelets), „Thrombos“ Normal: 150.000/µL-450.000/µL * <20.000/µL 28 % 20.000 bis 99.000/µL 47 % >100.000/µL 25 % * Die Normwerte für das Kindesalter stellen einen ungefähren Wertebereich dar. Bei exakter Anwendung von Normbereichen müssen diese nach Altersstufen (Neugeborene, Säuglinge, Kleinkinder, Schulkinder, Jugendliche) differenziert werden. Dies gilt vor allem für die Konzentration des Hämoglobins und die Anzahl der Leukozyten. Zur Veranschaulichung der Zusammenhänge wird auf diese Differenzierung verzichtet Tabelle 3. Laboruntersuchungsbefunde bei Erwachsenen mit akuter lymphatischer Leukämie[7] Laboruntersuchungsbefunde Wertebereich Prozent der Patienten Leukozyten; WBC (white blood cells), „Leukos“ Normal: 4.000/µL-11.000/µL * <5.000/µL 27 % 5.000 bis 10.000/µL 14 % >10.000/µL 59 % Hämoglobin; HGB, Hb Normal: 11,0–16,0 g/dL (Frauen), 13,0–18,0 (Männer) * <8,0 g/dL 28 % 8,0 bis 12,0 g/dL 51 % >12,0 g/dL 21 % Thrombozyten; PLT (platelets), „Thrombos“ Normal: 150.000/µL-450.000/µL * <25.000/µL 30 % 25.000 bis 150.000/µL 55 % >150.000/µL 15 % * Die Normwerte für Erwachsen variieren je nach Labor etwas. Bei Diagnosestellung hatten 92 % der Patienten Blasten im peripheren Blut. Diagnostik und Klassifikation
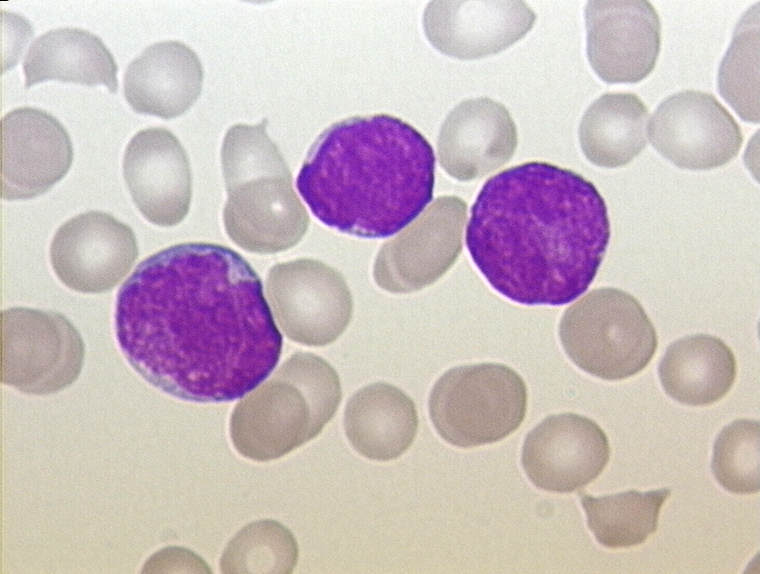
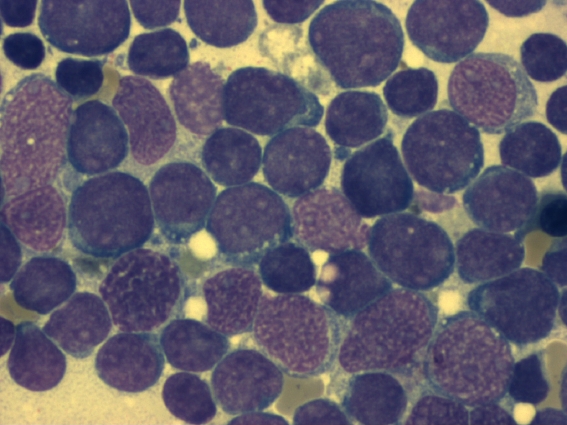
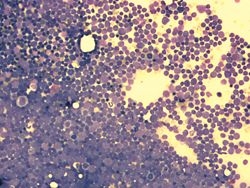 Knochenmarkausstrich bei ALL: mehr als 90 % der hier sichtbaren Zellen sind lymphatische Blasten (Leukämiezellen). Die Zelldichte ist stark gesteigert.
Knochenmarkausstrich bei ALL: mehr als 90 % der hier sichtbaren Zellen sind lymphatische Blasten (Leukämiezellen). Die Zelldichte ist stark gesteigert.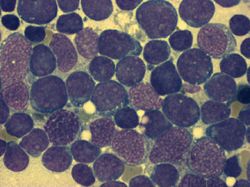 Knochenmarkausstrich bei ALL (Detail) mit Lymphoblasten (Leukämiezellen). Die Blasten weisen meist mehrere Nucleoli auf (Malignitätskriterium).
Knochenmarkausstrich bei ALL (Detail) mit Lymphoblasten (Leukämiezellen). Die Blasten weisen meist mehrere Nucleoli auf (Malignitätskriterium).Für die Diagnosestellung ist eine Untersuchung des Knochenmarks essentiell, da es vorkommen kann, dass bei Diagnosestellung noch keine feststellbare Ausschwemmung von Leukämiezellen aus dem Knochenmark in das Blut vorliegt. Man spricht dann von einer aleukämischen Verlaufsform (vgl. Tabellen 2 und 3). Wenn Leukämiezellen im Blut nachweisbar sind, aber die Zahl der Leukozyten insgesamt nicht erhöht ist, spricht man von einem subleukämischen Verlauf. Wenn die Gesamtzahl der Leukozyten durch Leukämiezellen erhöht ist, nennt man dies einen leukämischen Verlauf. Eine normale oder sogar erniedrigte Leukozytenzahl im Blut schließt also eine Leukämie nicht aus, entscheidend ist die Knochenmarkdiagnostik.
Die Diagnose einer ALL kann gestellt werden durch:
- Nachweis eines Anteils lymphatischer Blasten von mindestens 20 % bis 25 % im Knochenmark
- Zuordnung der Blasten zur lymphatischen Reihe durch Immunphänotypisierung (s. u.)
- Nachweis charakteristischer genetischer Veränderungen (s. u.)
Zytomorphologie
Durch mikroskopische Untersuchung des Knochenmarks oder des Blutausstrichs bei leukämischem Verlauf kann die Diagnose einer akuten Leukämie gestellt werden. Für die weitere Subklassifikation der ALL spielt die Beurteilung der Zellmorphologie im Gegensatz zur AML nur eine untergeordnete Rolle. In der sogenannten FAB-Klassifikation („French-American-British“) wird zwischen drei verschiedenen Morphologien unterschieden (L1, L2, L3). Von Bedeutung ist das nur für den seltenen L3-Subtyp, der mit der „reifzelligen B-ALL“ assoziiert ist. Die reifzellige B-ALL ist eine Sonderform der ALL und kann als die leukämische Manifestation des Burkitt-Lymphoms betrachtet werden (d. h. ein Burkitt-Lymphom mit >20 % oder >25 % Knochenmarkbefall) und wird wie dieses behandelt. Die Unterscheidung zwischen L1 und L2-Morphologie ist dagegen schwierig und selbst sehr erfahrene Hämatologen oder Hämatopathologen kommen hier zu unterschiedlichen Einschätzungen. Klinisch hat die Unterscheidung von L1 und L2 keine Bedeutung.
Tabelle 4. Häufigkeit der zytomorphologischen Befunde nach FAB-Klassifikation bei Kindern mit ALL[8] Zytomorphologische FAB-Klasse Charakteristika Prozent der Patienten FAB L1 Kleine Zellen vorwiegend mit einheitlicher Größe, gleichförmiges Chromatin, Zellkern nicht sichtbar oder klein, regelmäßig vorfindbare Nucleoli, sehr wenig Zytoplasma, leichte bis mäßige Basophilie, unterschiedliches Ausmaß von Vakuolen im Zytoplasma 84 % FAB L2 Große Zellen mit unterschiedliche Größe, ungleichförmiges Chromatin, Zellkern mit unregelmäßigen Spalten und Kerben, einer oder mehrere oftmals große Nucleoli, variable Größe des Zytoplasmas, mäßige bis oft starke Basophilie, unterschiedliches Maß an Vakuolen im Zytoplasma 15 % FAB L3 Große Zellen mit einheitlicher Größe, gleichförmig fein gesprenkeltes Chromatin, ovaler bis runder Zellkern, hervorstechende Nucleoli (auch mehrere), viel Zytoplasma, sehr starke Basophilie, oft hervorstechende Vakuolen im Zytoplasma 1 % Immunphänotypisierung
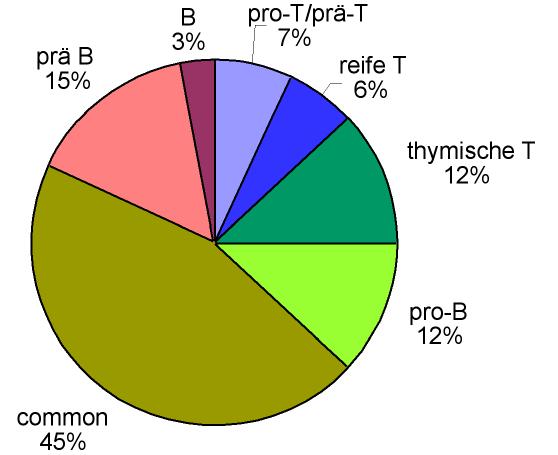
 Relative Häufigkeit der verschiedenen Immunphänotypen bei der ALL im Erwachsenenalter (Zahlen beruhen zum Teil auf Schätzungen), blau und grün dargestellt die T-Linien-ALL, rot, orange und gelb die B-Linien-ALL
Relative Häufigkeit der verschiedenen Immunphänotypen bei der ALL im Erwachsenenalter (Zahlen beruhen zum Teil auf Schätzungen), blau und grün dargestellt die T-Linien-ALL, rot, orange und gelb die B-Linien-ALLEntscheidend ist die Immunphänotypisierung der aus dem Blut oder mittels Knochenmarkpunktion gewonnenen Leukämiezellen, die heute meist mit dem FACS (Fluorescence Activated Cell Sorter) erfolgt. Mittels Immunphänotypisierung wird untersucht, ob und in welchem Ausmaß sich bestimmte Proteine an der Oberfläche oder im Zytoplasma der Zellen befinden. Das Expressionsmuster verschiedener lymphatischer, myeloischer und Vorläuferzell-Antigene ermöglicht die Zuordnung zu B- oder T-Zellreihe und die Festlegung des Differenzierungsstadiums. Man kann die ALL dementsprechend als B-Linien-ALL (mit B-lymphozytärer Differenzierung, weiter unterteilbar in „B-Vorläufer-ALL“ und „reif(zellig)e B-ALL“) oder T-Linien-ALL (mit T-lymphozytärer Differenzierung, weiter unterteilbar in „T-Vorläufer-ALL“ und „reife T-ALL“) einordnen. Tabelle 5 zeigt die Einteilung der akuten lymphatischen Leukämie aufgrund des Oberflächen-Antigenmusters nach der sogenannten EGIL-Klassifikation (EGIL = European Group for the Immunological Characterization of Leukemias)[9]. Die ALL-Zellen werden dabei nach ihrem „Reifungsgrad“ eingeteilt, d. h. eine pro-B-ALL ist eine ganz unreife Form, dann kommt die common ALL, gefolgt von der prä-B-ALL. Die reife B-ALL zeigt in ihrem Antigenmuster schon viele Merkmale reifer B-Lymphozyten. Analog ist es bei der T-Reihe: pro-T → prä T → thymische (corticale) T → reife T. Etwa 75 % der ALLs im Erwachsenenalter lassen sich der B-lymphozytären Reihe zuordnen und etwa 25 % der T-lymphozytären Reihe. Bei ALL im Kindes- und Jugendalter beträgt das Verhältnis etwa 85 % : 15 %.
Tabelle 5. Immunologische Klassifikation akuter lymphatischer Leukämien[9] B-Linien-ALL T-Linien-ALL pro-B common prä-B reife pro-T prä-T kortikale (thymische) reife B-Zell-Antigene CD19 + + + + - - - - cyCD22 + + + + - - - - CD79alpha + + + + - - - - cyIgM - - + - - - - - mIg - - - + - - - - T-Zell-Antigene cyCD3 - - - - + + +/- - CD7 - - - - + + + + CD2 - - - - - + + + CD1a - - - - - - + - mCD3 - - - - - - +/- + Vorläuferzell-Antigene TdT + + + - + + + +/- HLA-DR + + + + +/- - - - CD10 - + +/- +/- +/- +/- +/- - Zytogenetik und Molekulargenetik
Die Zytogenetik und Molekulargenetik der akuten Leukämien ist Gegenstand intensiver Forschungen. In den letzten zwei Jahrzehnten sind sehr viele Arbeiten erschienen, die sich mit verschiedenen Aspekten der Genetik dieser Erkrankungen beschäftigen. Die Genetik bildet gewissermaßen die Grundlage für das vertiefte Verständnis der Erkrankung.
Grundsätzlich ist zu sagen, dass sich die ALL des Kindes- oder Säuglingsalters, was die genetischen Grundlagen angeht, zum Teil erheblich von der ALL des Erwachsenenalters unterscheidet. Deswegen sollen die drei Altersklassen hier getrennt betrachtet werden:
ALL im Kleinkind-/Säuglingsalter (Alter unter einem Jahr)
Kennzeichnend für die ALL des Kleinkindes (engl.: infant leukemia) sind Veränderungen des MLL-Gens auf dem langen Arm von Chromosom 11, Bande 23 („11q23-Aberrationen“).[10] Diese Veränderungen sind mit einer ungünstigen Prognose assoziiert. Das MLL-Gen ist dabei in der Regel durch Chromosomentranslokationen mit anderen Genen fusioniert. Bis heute sind über 50 Fusionspartner bekannt. Die bei weitem häufigste Aberration ist die Fusion mit dem AF4-Gen auf Chromosom 4 (seltener auch ENL-Gen auf Chromosom 19, AF9-Gen auf Chromosom 9). Dabei entsteht ein sogenanntes Fusionsprotein („chimäres“ Protein), so dass die eigentlichen Funktionen der beteiligten Gene entweder verloren gehen oder so stark verändert werden, dass sie krankheitsauslösend oder -fördernd wirken.
ALL im Kindesalter (1 bis 15 Jahre)
Bei der ALL des Kindesalters sind eine Vielzahl von genetischen Veränderungen gefunden worden.[11] Vereinfacht unterscheidet man genetisch zur Zeit die folgenden Gruppen:
-
- Patienten mit hyperdiploidem Chromosomensatz (d. h. Vermehrung von Chromosomen) – prognostisch günstig
- Patienten mit hypodiploidem Chromosomensatz (d. h. Verminderung von Chromosomen) – prognostisch ungünstig
- Patienten mit der Chromosomentranslokation t(12;21), die zu dem Fusionsgen TEL-AML1 führt – prognostisch günstig [12]
- Patienten mit der Chromosomentranslokation t(9;22), die zu dem Fusionsgen BCR-ABL führt – prognostisch ungünstig
- Patienten mit der Chromosomentranslokation t(1;19), die zu dem Fusionsgen E2A-PBX1 führt – prognostisch günstig
- Patienten mit MLL-Aberrationen (s. o.) – prognostisch ungünstig
- Patienten mit T-Linien-ALL – prognostisch ungünstig
Die häufigste Veränderung ist dabei die t(12;21), die in etwa 25 % der Fälle zu finden ist.
ALL im Erwachsenenalter (ab 16 Jahre)
Bei Erwachsenen findet man im Prinzip dieselben genetischen Veränderungen wie bei Kindern, nur mit zum Teil erheblich unterschiedlicher Häufigkeit. Die häufigste Veränderung ist dabei die t(9;22), die in ca. 25 % der Fälle zu finden ist. Die t(12;21) ist dagegen selten (ca. 1 %). Die prognostische Wertigkeit der o. g. Veränderungen ist bei Erwachsenen z. T. noch nicht eindeutig geklärt (t(12;21), t(1;19)). Die T-ALL hat eine bessere Prognose als bei Kindern.
Behandlung
Prognostische Faktoren
Die ALL ist kein einheitliches Krankheitsbild sondern kann bei verschiedenen Patienten einen sehr unterschiedlichen Verlauf nehmen. Manche Patienten zeigen z. B. ein gutes Therapieansprechen während andere nur verzögert auf die Therapie ansprechen oder schnell rezidivieren. In den letzten Jahrzehnten konnten verschiedene Risikofaktoren identifiziert werden. Als Risikofaktoren bezeichnet man in diesem Kontext Faktoren, die dazu führen, dass der betroffene Patient ein erhöhtes Risiko aufweist, schlecht auf die Behandlung anzusprechen oder ein Rezidiv zu erleiden.
Die ALL wird weltweit nicht einheitlich behandelt. Es gibt in verschiedenen Ländern verschiedene große sogenannte Studiengruppen, die ihre Patienten nach bestimmten Therapieschemata behandeln. Mittlerweile gibt es aber Bestrebungen, die Therapien aneinander anzugleichen oder länderübergreifende Therapiestudien auf den Weg zu bringen (z. B. im Rahmen des European LeukemiaNet).
Beispiele für solche Studiengruppen sind:
Studiengruppen für erwachsene Patienten
-
- GMALL (German Multicenter ALL Study Group) – in Deutschland und einigen weiteren Ländern
- GIMEMA (Gruppo Italiano Malattie Ematologiche dell’ Adulto) – in Italien
- MRC (Medical Research Counsil) – im Vereinigten Königreich
- PETHEMA (Programa para el Estudio de la Terapéutica en Hemopatía Maligna) – in Spanien
- GRAALL (Group for Research on Adult Acute Lymphoblastic Leukemia) – in Frankreich, Belgien und der Schweiz
- NILG (Northern Italy Leukaemia Group) – in Norditalien
- PALG (Polish Adult Leukaemia Group) – in Polen
- SWOG (Southwest Oncology Group) – in den USA
Studiengruppen für Kinder und Jugendliche
-
- ALL-BFM Berlin-Frankfurt-Münster kooperative multizentrische ALL Studie – für primäre Erkrankung – Deutschland, Österreich, Schweiz, z. T. auch andere europäische Länder
- COALL Cooperative ALL-Studie – für primäre Erkrankung – Deutschland
- ALL-REZ BFM Berlin-Frankfurt-Münster kooperative multizentrische Studie ALL-Rezidiv – für Rückfall – Deutschland, Österreich, Schweiz
- ALL-SZT BFM Berlin-Frankfurt-Münster kooperative multizentrische Studie ALL-Stammzelltransplantation – Deutschland, Österreich, Schweiz
- I-BFM (International BFM-Group) – Behandlungsprotokoll nach Grundlage von ALL-BFM – Europa
- COG (Children’s Oncology Group) – diverse Protokolle – USA
- UKALL (United Kingdom ALL Study Group) – Behandlungsprotokoll der UK Children’s Cancer Study Group – Großbritannien
- SFOP (Societé française d’oncologie pédiatrique) – Frankreich
- NOPHO (Nordic Society of Paediatric Haematology and Oncology) – Skandinavien
Etablierte Risikofaktoren
Fast alle Studiengruppen haben die folgenden Risikofaktoren herausarbeiten können – obwohl die jeweilige Therapie unterschiedlich ist:
- 1. genetisch Nachweis des BCR-ABL-Fusionsgens [13], oft (aber nicht immer) zytogenetisch sichtbar als „Philadelphia-Chromosom“
- 2. hohe periphere Leukozytenzahl bei Diagnosestellung als Ausdruck einer hohen „Tumorlast“ (tumor burden)
- 3. verzögertes Ansprechen auf die Therapie (insbesondere nach Induktionsphase der Therapie)
- 4. genetisch Nachweis eines MLL-Fusionsgens
- 5. das Patientenalter: junge Patienten haben in der Regel deutlich bessere Heilungschancen als ältere (Ausnahme: Säuglinge)[14]
- 6. Befall des Zentralnervensystems (Gehirn und Rückenmark) durch die ALL [15][16]
- 7. bestimmte Immunphänotypen (z. B. T-Linien ALL im Kindes- und Jugendalter (1–18 Jahre)[17]
Andere Risikofaktoren sind umstritten und nicht allgemein akzeptiert. Es muss auch betont werden, dass die oben genannten Faktoren statistische Risikofaktoren sind, d. h. im Einzelfall kann der klinische Verlauf auch anders aussehen, als erwartet. Die Identifizierung von Riskofaktoren ist deswegen bedeutsam, weil es sich bei den betroffenen Patienten um Hochrisko-Patienten handelt, die sehr gefährdet sind, ein Rezidiv zu erleiden. Deswegen sehen die bisherigen Therapiekonzepte für diese Patienten primär eine intensivere Behandlung vor. In der Regel wird bei Erwachsenen die allogene Knochenmark- oder Stammzelltransplantation angestrebt.
Minimale Resterkrankung
Einen besonderen Risikofaktor, der hier gesondert abgehandelt werden soll, stellt die sogenannte „minimale Resterkrankung“ (MRD = minimal residual disease) dar. Als MRD bezeichnet man den Anteil von Leukämiezellen nach einer Therapie. Man spricht dann z. B. von einer MRD von 0,001 % oder 10-5, was dann eine Leukämiezelle unter 100.000 gesunden Blutzellen bezeichnet. Man weiß mittlerweile, dass die MRD den wichtigsten Risikofaktor bei der Leukämiebehandlung überhaupt darstellt. Im Rahmen moderner Leukämie-Therapiekonzepte wird angestrebt, die MRD deswegen auch regelmäßig zu bestimmen, um den Grad des Therapieansprechens zu ermitteln. Meist wird die MRD mittels Polymerase-Kettenreaktion (PCR) ermittelt. Dazu sind hochspezialisierte und in dieser speziellen Diagnostik erfahrene Labore erforderlich. Gelingt es durch die Therapie nicht, die MRD unter ein gewisses Niveau zu bringen (mindestens 10-4), ist ein Rezidiv der Erkrankung praktisch vorbestimmt.[18][19][20][21] In neueren Therapiestudien ist die Behandlung wesentlich MRD-basiert „stratifiziert“, d. h. bei den behandelten Patienten wird regelmäßig die MRD gemessen und je nach der „Kinetik“, d. h. dem MRD-Verlauf werden diese Patienten unterschiedlichen weiteren Behandlungsformen zugeführt.
Chemotherapie
Zur Behandlung der ALL können verschiedene Chemotherapie-Verfahren eingesetzt werden.
Vor Beginn der Behandlung muss eine ausführliche Diagnostik erfolgen, um die ALL genetisch und immunzytolologisch zu charakterisieren (s. o.). Dazu gehört auch eine Untersuchung des Liquor cerebrospinalis durch Lumbalpunktion und eventuell Bildgebung des Zentralnervensystems (beispielsweise durch Magnetresonanztomographie). Unmittelbar anschließend an die Diagnostik wird die Behandlung begonnen. Diese erfolgt mittels Chemotherapie, d. h. durch Gabe von Zytostatika. Es werden Kombinationen von verschiedenen Zytostatika gegeben, da dadurch die antileukämische Wirkung vielfach verstärkt ist. Die Behandlung dauert insgesamt mindestens 12 bis 24 Monate.
Vereinfacht kann man sagen, dass die Behandlung nach folgendem Schema abläuft:
Induktionstherapie (u. U. als „Doppel-Induktion“) → Konsolidierungstherapie → Re-Induktionstherapie → Erhaltungstherapie
Induktionsphase (1–2 Monate)
Am Anfang steht die „Induktionstherapie“, die sehr intensiv und nebenwirkungsreich ist. Die wichtigsten Zytostatika sind in dieser Phase
-
- Anthracycline, z. B. Daunorubicin oder Doxorubicin
- Antimetaboliten, z. B. Cytarabin
- Kortikosteroide, z. B. Dexamethason und Prednison (gehört nicht zur Gruppe der Zytostatika, Kortison hat eine entzündungshemmende und immunsuppriemierend Wirkung)
- Vinca-Alkaloide, z. B. Vincristin
- Alkylantien, z. B. Cyclophosphamid
- L-Asparaginase.
An genau festgelegten Tagen des Behandlungsablaufes werden dem Patienten definierte Zytostatika in vorbestimmten Dosierungen und Kombinationen verabreicht. Die Induktionsphase dauert 4 bis 6 Wochen (bei unkompliziertem Verlauf). Sie hat das Ziel, die ALL so zurückzudrängen, dass sie am Ende der Induktionsphase (ca. einen Monat nach Behandlungsbeginn) in der Knochenmarkpunktion nicht mehr nachweisbar ist. Die Behandlung ist deswegen so intensiv, weil man den Blasten keine Zeit geben möchte Resistenzen gegen Zytostatika zu entwickeln und deswegen eine möglichst rasche Reduktion der Tumorlast erzielen will.
Gelegentlich wird nach der ersten Induktionstherapie noch eine zweite Induktionstherapie angeschlossen („Doppelinduktion“), die etwas andere Medikamentenkombinationen beinhaltet und auch etwa einen Monat dauert.
Konsolidierung (mehrere Monate)
Obwohl die ALL im Idealfall einen Monat nach Therapiebeginn nicht mehr nachweisbar sein kann, wird die Behandlung fortgesetzt. Eine Beendung der Behandlung zu diesem Zeitpunkt würde ansonsten mit sehr hoher Wahrscheinlichkeit zu einem Rückfall führen: die ALL ist zwar mit den zur Verfügung stehenden diagnostischen Methoden kaum oder nicht mehr nachweisbar, aber noch nicht beseitigt. An die Induktionsphase schließt sich die „Konsolidierungsphase“ an. Auch in dieser Phase werden Zytostatika in festgelegten Dosen und Zeitabständen verabreicht. Die wichtigsten Zytostatika in der Konsolidierungsphase sind:
-
- Methotrexat
- Cyclophosphamid
- Cytarabin
- Etoposid oder früher auch Teniposid
- L-Asparaginase
Insbesondere die Zytostatika Methotrexat und Cytarabin werden dabei in mittelhohen bis hohen Dosierungen eingesetzt. Zusätzlich erfolgt die zwischenzeitliche Gabe von Tioguanin und 6-Mercaptopurin.
Reinduktion
An die Konsolidierungsphase schließt sich die „Re-Induktionsphase“ an. In dieser wird analog zur Induktionsphase die Behandlung erneut intensiviert. Die dabei eingesetzten Zytostatika entsprechen denen der Induktionsphase.
Induktionsphase, Konsolidierungsphase und Re-Induktionsphase werden auch als Intensivphase der Behandlung bezeichnet. Die Gesamtdauer der Behandlung ist abhängig von den Risikofaktoren und dem Behandlungsverlauf der betroffenen Patienten. Die Dauer schwankt zwischen 6 und 12 Monaten. Ein stationärer Krankenhausaufenthalt ist in der Intensivphase zumeist bei der Verabreichung der Zytostatika und bei Auftreten von Komplikationen wie beispielsweise Infektionen in Knochenmarkaplasie („Zelltief“) erforderlich. Zwischen den einzelnen Zytostatika-Zyklen verweilt der Patient mit problemlosem Verlauf zu Hause; lediglich ambulante Kontrollen des Blutbildes sind erforderlich und sinnvoll.
Erhaltungstherapie
In der sich erneut unmittelbar an die Intensivphase anschließenden „Erhaltungsphase“ erhalten die Patienten eine Chemotherapie mit Methotrexat und 6-Mercaptopurin.[22] Im Gegensatz zu den vorhergehenden Behandlungsphasen wird die Chemotherapie in dieser Phase teilweise oder auch vollständig als orale Chemotherapie (in Form von Tabletten) durchgeführt: 6-Mercaptopurin wird einmal täglich, Methotrexat einmal wöchentlich eingenommen. Ein Krankenhausaufenthalt ist im Gegensatz zur Intensivphase nicht erforderlich. Die Dauer der Erhaltungsphase ist in Abhängigkeit vom verwendeten Therapieprotokoll unterschiedlich und beläuft sich auf 6–18 Monate. Im Kindes- und Jugendalter wird die Erhaltungstherapie mit 6-Mercaptopurin und Methotrexat typischerweise bis zum Zeitpunkt zwei Jahre nach Diagnosestellung bzw. Behandlungsbeginn durchgeführt.
ZNS-Prophylaxe, ZNS-Bestrahlung
Ein Problem der meisten Zytostatika ist der Umstand, dass sie gar nicht oder nur schlecht die Blut-Hirn-Schranke überwinden. Dringen Leukämiezellen in das Zentralnervensystem (ZNS) ein, sind sie dort vor Zytostatika geschützt und können von dort aus zu einem Rezidiv der Erkrankung führen. Um solche ZNS-Rezidive zu verhindern wird daher bei allen Patienten eine sogenannte ZNS-Prophylaxe durchgeführt. Diese kann in zwei Formen erfolgen. Zum einen als mehrfach wiederholte direkte Gabe von Zytostatika (i. d. R. Methotrexat) in den Liquorraum („Nervenwasser“) mittels Lumbalpunktion (= intrathekale Gabe), zum andern als Bestrahlung des Zentralnervensystems. Die ZNS-Bestrahlung war insbesondere zu Beginn der ALL-Behandlung Standard. Im Kindes- und Jugendalter ist die ZNS-Bestrahlung aufgrund der beobachteten Spätschäden für die meisten Patienten zusehends durch die intrathekale Gabe von Methotrexat ersetzt worden.[23] Bei einem gesicherten Befall des Zentralnervensystems zu Anfang der Behandlung oder bei Erfüllung bestimmter Risikofaktoren wie beispielsweise einer anfänglichen Leukozytenzahl von mehr als 100.000 pro Mikroliter im peripheren Blut wird eine Bestrahlung des Zentralnervensystems jedoch auch bei Kindern immer noch durchgeführt.[24][25]
Mediastinalbestrahlung
Wenn eine starke Thymusschwellung durch Leukämiezellen als sogenannter Mediastinaltumor vorliegt, wird diese bei Erwachsenen lokal bestrahlt (bei Kindern und Jugendlichen nur im absoluten Ausnahmefall, da der Thymus bei Kindern im Gegensatz zu Erwachsenen noch wesentliche Funktionen im Immunsytem erfüllt.).
Kontrolle des Therapieerfolges
In regelmäßigen Abständen wird eine erneute Knochenmarkpunktion durchgeführt um den Erfolg der Therapie zu überprüfen. Von besonderer Wichtigkeit ist dabei die Knochenmarkpunktion nach Abschluss der Induktionsphase (nach 4 bis 6 Wochen oder Tag 33 nach Behandlungsbeginn). Auch regelmäßige Überprüfungen des Nervenwassers (Liquor) auf das Vorhandensein von ALL-Zellen sind unabdingbar.
Die Behandlung kann nicht beliebig intensiv und hochdosiert sein, da die Zytostatika erhebliche Nebenwirkungen haben, die dosislimitierend sind. Die wichtigste Nebenwirkung ist die Knochenmarkdepression, d. h. die Schädigung auch der verbliebenen, beim Leukämiekranken ohnehin schon stark geschwächten gesunden Blutbildung. Als Folge der chemotherapeutischen Behandlung kommt es zu einer länger dauernden „Aplasie“, d. h. einem fast vollständigen Ausfall der normalen Blutbildung. In dieser Phase, die bis zu einem Monat und länger dauern kann, müssen fehlende Blutbestandteile durch Transfusionen ersetzt werden und der Patient ist aufgrund der fehlenden Immunabwehr extrem infektgefährdet. Nicht wenige Patienten versterben aufgrund schwerer in Aplasie erworbener Infektionen während der Behandlung. In der Regel wird durch die Induktionstherapie, die sich meist über etwa 2–3 Monate hinzieht (die Aplasie-Zeiten mitgerechnet) die „Leukämie-Tumorlast“ um mindestens einen Faktor 1.000 bis 10.000 oder mehr reduziert. In der nach Ende der Induktionsphase in der Regel nochmal durchgeführten Knochenmarkpunktion sollte keine Restleukämie mehr sichtbar sein (zum Begriff der Minimalen Resterkrankung siehe oben).
Insgesamt ist die Behandlung sehr nebenwirkungsreich und ist trotz aller ärztlichen Maßnahmen mit erheblichen Gefahren, insbesondere durch Infektionen aufgrund der geschwächten Immunabwehr verbunden. Eine große Hoffnung für die Zukunft bieten „targeted drugs“, d. h. Medikamente, die wesentlich spezifischer auf Leukämiezellen wirken als herkömmliche Zytostatika, und damit ein weniger schwerwiegendes Nebenwirkungsprofil haben. Ein Beispiel für eine solche Substanz ist der Wirkstoff Imatinib (Glivec®), der bei BCR-ABL-positiver ALL eingesetzt wird. Aber auch monoklonale Antikörper wie Rituximab oder Alemtuzumab werden die Therapie in Zukunft wesentlich verändern.
Behandlungsergebnisse
Bevor wirksame Zytostatika zur Verfügung standen, bedeutete die Diagnose „akute lymphatische Leukämie“ praktisch das Todesurteil für die betroffenen Patienten. Je nach Stadium der Erkrankung verstarben die Betroffenen innerhalb von Tagen bis Wochen nach Diagnosestellung. Haupttodesursachen waren schwere Infektionen aufgrund der schweren Immunschwäche, Spontanblutungen aufgrund der Thrombozytopenie, oder andere Komplikationen (z. B. bei Befall des Zentralnervensystems). Besonders tragisch und schwerwiegend war der Umstand, dass häufig auch kleine Kinder von der Erkrankung betroffen waren (die ALL ist die häufigste bösartige Erkrankung des Kindesalters). Noch Ende der 70er Jahre, als schon einige wirksame Zytostatika zur Verfügung standen, lag das mittlere 5-Jahres-Überleben bei erwachsenen ALL-Patienten in Deutschland bei unter 15 %. Bei Kindern waren die Ergebnisse besser. Seit mehreren Jahrzehnten sind intensive Anstrengungen gemacht worden, die Therapieergebnisse für ALL-Patienten zu verbessern. Dies geschah durch umfangreiche nacheinander abfolgende klinische Studien, in denen die Patienten behandelt wurden. Die Ergebnisse und Erfahrungen aus einer Studie dienten dann dazu, eine neue darauf folgende verbesserte Therapiestudie zu planen. Insgesamt liegt das Gesamtüberleben bei ALL-Patienten zur Zeit bei ca. 80 % bei Kindern und ca. 40–45 % bei Erwachsenen. Diese Zahlen variieren, da sie vom jeweiligen Subtyp und der Therapie abhängen. Weitere Verbesserungen sind in der Zukunft zu erwarten. Es muss aber betont werden, dass diese Zahlen Mittelwerte darstellen. Im Einzelfall kann die Prognose deutlich davon abweichen. So kann z. B. ein 20-jähriger Patient ohne Risikofaktoren (s. o.) deutlich bessere Heilungschancen erwarten, als z. B. ein 65-jähriger Patient mit Risikofaktoren.
Geschichtliches
Der Begriff „Leukämie“ wurde von Rudolf Virchow geprägt,[26] der 1845 zusammen mit und unabhängig von dem britischen Pathologen John Hughes Bennett [27] das Krankheitsbild einer chronischen myeloischen Leukämie beschrieb.[28] Nach dem Aufkommen synthetischer chemischer Farbstoffe entwickelte Paul Ehrlich 1877 Techniken zur Färbung von Blutausstrichen, die eine genauere Unterscheidung von Leukozyten ermöglichten. Wilhelm Ebstein prägte 1889 den Begriff "akute Leukämie" in Abgrenzung von den chronischen Leukämien.[29] Mosler beschrieb 1879 erstmals die Techniken der Knochenmark-Untersuchung zur Diagnose einer Leukämie.[30] 1900 führte Otto Naegeli die Einteilung der Leukämien in "myeloische" und "lymphatische" ein.[31]
Literatur
- Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med 2004; 350:1535–1548. PMID 15071128
- Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354:166–178. PMID 16407512
- Gökbuget N (Hrsg.). Akute lymphatische Leukämie. Verlag UNI-MED Science, 1. Auflage 2007, ISBN 978-3-89599-218-6
Einzelnachweise
- ↑ Surveillance, Epidemiology, and End Results (SEER) Program of the National Cancer Institute (NCI)
- ↑ Deutsches Kinderkrebsregister
- ↑ Die Vereinigung schweizerischer Krebsregister [1] enthält nur Daten aus ausgewählten Kantonen und hat, genau so wie Statistik Austria [2] in Österreich alle Leukämieerkrankungen zusammen erfasst, wovon die ALL natürlich nur einen Bruchteil ausmacht.
- ↑ Surveillance, Epidemiology, and End Results (SEER) Program of the National Cancer Institute (NCI)
- ↑ Margolin JF, Steuber CP, Poplack DG: Acute Lymphoblastic Leukemia. In: Pizzo PA, Poplack DG (Hrsg.): Principles and Practice of Pediatric Oncology. 4th Edition. Lippincott Williams & Wilkins. Baltimore, 2003. ISBN 0-7817-2658-1
- ↑ Margolin JF, Steuber CP, Poplack DG: Acute Lymphoblastic Leukemia. In: Pizzo PA, Poplack DG (Hrsg.): Principles and Practice of Pediatric Oncology. 4th Edition. Lippincott Williams & Wilkins. Baltimore, 2003. ISBN 0-7817-2658-1
- ↑ Hoelzer D, Gökbuget N: Akute lymphatische Leukämie. In: Seeber S., Schütte J. (Hrsg.) „Therapiekonzepte Onkologie“. Springer-Verlag Heidelberg, 2007, S. 271. ISBN 978-3-540-28588-5
- ↑ Margolin JF, Steuber CP, Poplack DG: Acute Lymphoblastic Leukemia. In: Pizzo PA, Poplack DG (Hrsg.): Principles and Practice of Pediatric Oncology. 4th Edition. Lippincott Williams & Wilkins. Baltimore, 2003. ISBN 0-7817-2658-1
- ↑ a b Bene MC et al. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia. 1995;9(10):1783–6. PMID 7564526
- ↑ Jansen MW, et al.: Immunobiological diversity in infant acute lymphoblastic leukemia is related to the occurrence and type of MLL gene rearrangement. Leukemia. 2007;21:633–41. PMID 17268512
- ↑ Forestier E et al. Cytogenetic findings in a population-based series of 787 childhood acute lymphoblastic leukemias from the Nordic countries. The NOPHO Leukemia Cytogenetic Study Group. Eur J Haematol 2000; 64(3):194–200. PMID 10997816
- ↑ Loh ML et al. Prospective analysis of TEL/AML1-positive patients treated on Dana-Farber Cancer Institute Consortium Protocol 95–01. Blood 2006; 107(11):4508–4513. PMID 16493009
- ↑ Ribera JM et al. Prognostic value of karyotypic analysis in children and adults with high-risk acute lymphoblastic leukemia included in the PETHEMA ALL-93 trial. Haematologica 2002; 87(2):154–166. PMID 11836166
- ↑ Möricke A et al. Prognostic impact of age in children and adolescents with acute lymphoblastic leukemia: data from the trials ALL-BFM 86, 90, and 95. Klin Padiatr 2005; 217:310–320. PMID 16307416
- ↑ te Loo DM et al. Prognostic significance of blasts in the cerebrospinal fluid without pleiocytosis or a traumatic lumbar puncture in children with acute lymphoblastic leukemia: experience of the Dutch Childhood Oncology Group. J Clin Oncol 2006; 24(15):2332–2336. PMID 16710032
- ↑ Clarke M et al. CNS-directed therapy for childhood acute lymphoblastic leukemia: Childhood ALL Collaborative Group overview of 43 randomized trials. J Clin Oncol 2003; 21(9):1798–1809. PMID 12721257
- ↑ Goldberg JM et al. Childhood T-cell acute lymphoblastic leukemia: the Dana-Farber Cancer Institute acute lymphoblastic leukemia consortium experience. J Clin Oncol. 2003;21(19):3616–22. PMID 14512392
- ↑ Marshall GM et al. Importance of minimal residual disease testing during the second year of therapy for children with acute lymphoblastic leukemia. J Clin Oncol 2003; 21(4):704–709. PMID 12586809
- ↑ Nyvold C et al. Precise quantification of minimal residual disease at day 29 allows identification of children with acute lymphoblastic leukemia and an excellent outcome. Blood 2002; 99(4):1253–1258. PMID 11830473
- ↑ Brüggemann M et al. Clinical significance of minimal residual disease quantification in adult patients with standard-risk acute lymphoblastic leukemia. Blood. 2006;107(3):1116–23. PMID 16195338
- ↑ Raff T et al. Molecular relapse in adult standard-risk ALL patients detected by prospective MRD monitoring during and after maintenance treatment: data from the GMALL 06/99 and 07/03 trials. Blood. 2007;109(3):910–5. PMID 17023577
- ↑ Harms DO et al. Thioguanine offers no advantage over mercaptopurine in maintenance treatment of childhood ALL: results of the randomized trial COALL-92. Blood 2003; 102(8):2736–2740. PMID 12843002
- ↑ Langer T et al. CNS late-effects after ALL therapy in childhood. Part III: neuropsychological performance in long-term survivors of childhood ALL: impairments of concentration, attention, and memory. Med Pediatr Oncol 2002; 38(5):320–328. PMID 11979456
- ↑ Laver JH et al. Effects of cranial radiation in children with high risk T cell acute lymphoblastic leukemia: a Pediatric Oncology Group report. PMID 10720128
- ↑ Conter V et al. Role of cranial radiotherapy for childhood T-cell acute lymphoblastic leukemia with high WBC count and good response to prednisone. Associazione Italiana Ematologia Oncologia Pediatrica and the Berlin-Frankfurt-Münster groups. Leukemia. 2000;14(3):369–73. J Clin Oncol. 1997;15(8):2786–91. PMID 9256120
- ↑ Virchow R: Die Leukämie. In Virchow R (ed): Gesammelte Abhandlungen zur Wissenschaftlichen Medizin. Frankfurt, Meidinger, 1856, S. 190.
- ↑ Bennett, J.H.: Case of hypertrophy of the spleen and liver, in which death took place from suppuration of the blood. Edinburgh Medical and Surgical Journal, 1845,64:413-23.
- ↑ Virchow, R: Weisses Blut. Froriep’s Notizen aus dem Gebiete der Natur- und Heilkunde, 1845;36:151-6.
- ↑ Ebstein W. Über die acute Leukämie und Pseudoleukämie. Deutsch Arch Klin Med. 1889,44:343.
- ↑ Mosler F. Klinische Symptome und Therapie der medullären Leukämie. Berl Klin Wochenschr. 1876,13:702.
- ↑ Naegeli O. Über rothes Knochenmark und Myeloblasten. Deutsch Med Wochenschr. 1900,26:287.
Weblinks
- Kompetenznetz „Akute und chronische Leukämien“
- Kompetenznetz „Pädiatrische Onkologie und Hämatologie“
- European LeukemiaNet mit Links zur Studiengruppen und Behandlungsprotokollen
- Southwest Oncology Group (SWOG) in den USA
- Audio-Beitrag zu akuten Leukämien
Wikimedia Foundation.
Schlagen Sie auch in anderen Wörterbüchern nach:
Akute lymphatische Leukämie — Klassifikation nach ICD 10 C91.0 Akute lymphoblastische Leukämie ICD O 9835/3 (B Linie) ICD O 9837/3 (T Linie) … Deutsch Wikipedia
6-TG — Strukturformel Allgemeines Freiname Tioguanin Andere Namen 6 … Deutsch Wikipedia
6-Thioguanin — Strukturformel Allgemeines Freiname Tioguanin Andere Namen 6 … Deutsch Wikipedia
6TG — Strukturformel Allgemeines Freiname Tioguanin Andere Namen 6 … Deutsch Wikipedia
Thioguanin — Strukturformel Allgemeines Freiname Tioguanin Andere Namen 6 … Deutsch Wikipedia
Krebserkrankungsarten — Die Internationale statistische Klassifikation der Krankheiten und verwandter Gesundheitsprobleme (ICD 10) klassifiziert in ihrem Kapitel II Neubildungen (Tumoren) nach untenstehendem Schlüssel. Als Weiterentwicklung des ICD 10 speziell für die… … Deutsch Wikipedia
Liste der Neubildungen nach ICD-10 — Die Internationale statistische Klassifikation der Krankheiten und verwandter Gesundheitsprobleme (ICD 10) klassifiziert in ihrem Kapitel II Neubildungen (Tumoren) nach untenstehendem Schlüssel. Als Weiterentwicklung des ICD 10 speziell für die… … Deutsch Wikipedia
Tioguanin — Strukturformel Allgemeines Freiname Tioguanin Andere Namen … Deutsch Wikipedia
Alexan — Strukturformel Allgemeines Freiname Cytarabin Andere Namen 1 β … Deutsch Wikipedia
Ara-C — Strukturformel Allgemeines Freiname Cytarabin Andere Namen 1 β … Deutsch Wikipedia
 18+© Academic, 2000-2026
18+© Academic, 2000-2026- Kontaktieren Sie uns: Unterstützung, Werbung
Wörterbücher Export, schritte mit PHP, Joomla, Drupal, WordPress, MODx.
- Störungen der Blutbildung: