- Akute myeloische Leukämie
-
Klassifikation nach ICD-10 C92.0 Akute myeloische Leukämie ICD-10 online (WHO-Version 2011) Die akute myeloische Leukämie (AML) ist eine maligne (bösartige) Erkrankung des blutbildenden Systems und zwar der Myelopoese, also des Teils des blutbildenden Systems, der für die Bildung von Granulozyten, Monozyten, Erythrozyten und Megakaryozyten verantwortlich ist. Sie führt zu einer z. T. massiven Vermehrung unreifer Vorstufen der Myelopoese im Knochenmark und in der Mehrzahl der Fälle auch im Blut (Leukozytose).
Der Begriff „Leukämie“ wurde 1845 von Rudolf Virchow geprägt, der damals das Krankheitsbild einer chronischen myeloischen Leukämie beschrieb. Die Entwicklung von Färbeverfahren für Blutausstriche im Jahre 1891 durch Paul Ehrlich führte zu neuen Erkenntnissen über die Morphologie der akuten Leukämien und ermöglichte in der Folge die Abgrenzung der myeloischen von der lymphatischen Leukämie (Naegeli 1900).Inhaltsverzeichnis
Epidemiologie
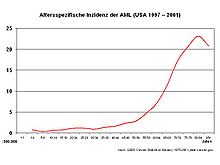
Die AML ist eine seltene Erkrankung mit einer Inzidenz von etwa drei Neuerkrankungen/100.000 im Jahr. In Deutschland treten etwa 3.600 Neuerkrankungen pro Jahr auf. Sie ist überwiegend eine Erkrankung des höheren Lebensalters, das mediane Alter bei Diagnosestellung liegt bei 63 Jahren. Die AML macht etwa 80 % aller akuten Leukämien bei Erwachsenen aus. Männer sind etwas häufiger betroffen als Frauen (Verhältnis 1,4:1). Im Kindesalter haben nur 15 bis 20 % der Patienten mit einer akuten Leukämie eine AML. Allerdings handelt es sich bei der seltenen akuten Leukämie im Neugeborenenalter meist um eine AML.
Ursachen und Entstehung
Bekannte Risikofaktoren für die Entwicklung einer AML sind eine Exposition gegenüber einer hohen Dosis ionisierender Strahlung (z. B. nach den Atombombenexplosionen in Hiroshima und Nagasaki) und eine langjährige chronische Belastung mit Benzol. Auch nach Anwendung bestimmter Zytostatika, wie Alkylantien und Etoposid, kann es nach einer Latenzzeit von mehreren Jahren zur Entwicklung einer AML kommen. Eine AML tritt gehäuft bei einigen genetischen Erkrankungen wie z. B. dem Down-Syndrom auf. Auch das Rauchen spielt eine Rolle bei der Entstehung der AML. In vielen Fällen bleibt die Ursache jedoch unklar.
Die Mechanismen, die zur Entwicklung einer AML führen, sind Gegenstand der aktuellen Forschung. Man geht heute davon aus, dass am Anfang der Leukämieentstehung genetische Veränderungen in einer einzelnen hämatopoetischen Vorläuferzelle stehen. Diese Veränderungen führen zu einer klonalen Vermehrung unreifer Zellen, die die Fähigkeit zur Ausreifung verloren haben. Bei zahlreichen bei der AML bekannten chromosomalen Aberrationen (s.u. Zytogenetik) sind häufig Gene involviert, die in der normalen Zellregulation eine wichtige Rolle spielen. Durch Translokationen entstehen z. T. neue Fusionsgene, die an der Leukämieentstehung beteiligt sind, z. B. PML/RARα bei t(15;17), AML1/ETO bei t(8;21).
Klinik und Verlauf
Die leukämischen Zellen breiten sich in Knochenmark und Blut aus, und können auch Lymphknoten, Milz sowie weitere Organe, in seltenen Fällen auch das ZNS infiltrieren. Unmittelbare Folge ist eine Verdrängung der normalen Blutbildung (Hämatopoese). Es entsteht ein Mangel an Erythrozyten, Blutplättchen und funktionsfähigen reifen Granulozyten.
Die Symptome bei AML sind vorwiegend auf die Knochenmarkinsuffizienz zurückzuführen. Es handelt sich häufig um ein akutes Krankheitsbild, typische Symptome sind:
- Allgemeine Schwäche, Krankheitsgefühl, Blässe
- Nachtschweiß
- Zeichen der Blutgerinnungsstörung mit Petechien, Hämatomen oder Schleimhaut- oder Zahnfleischblutungen
- Infektionen unterschiedlicher Lokalisation wie z. B. Pneumonie („Lungenentzündung“), Tonsillitis („Mandel-Entzündung“) oder unklares Fieber
- Entzündung der Mundschleimhaut, Mundsoor
- Eher selten mäßige Milzvergrößerung und/oder Lymphknotenschwellungen, gelegentlich Zahnfleischschwellung (Gingiva-Hyperplasie), diese Veränderungen kommen vor allem bei der monozytären Leukämie vor
Häufig auftretende weitere Befunde sind:
- In den meisten Fällen Leukozytose, manchmal auch normale oder sogar erniedrigte Leukozytenzahl
- Auftreten von Blasten im Differentialblutbild
- Anämie und Thrombopenie
- Erhöhung von LDH, BSG und Harnsäure
- Gerinnungsstörungen (vorwiegend bei Promyelozyten-Leukämie oder monozytärer Leukämie)
Unbehandelt schreitet die Erkrankung schnell voran und führt nach einigen Wochen zum Tode, meist aufgrund von unbeherrschbaren Infektionen oder Blutungen.
Diagnostik und Klassifikation
Die Verdachtsdiagnose einer akuten Leukämie ergibt sich aus den klinischen Symptomen und dem Blutbild einschließlich Differentialblutbild. Die Diagnose wird gesichert durch eine Untersuchung des Knochenmarks (siehe Knochenmarkpunktion). Entscheidend für die Behandlung und Prognose ist die Abgrenzung einer AML von einer Akuten lymphatischen Leukämie (ALL).
Die moderne Diagnostik beruht auf der Kombination morphologischer und zytochemischer Befunde, ergänzt durch die Immunphänotypisierung und Zytogenetik und ggf. molekulare Diagnostik. In Zukunft wird vermutlich die Analyse von Genexpressionsprofilen mittels der Microarray-Technik zunehmende Bedeutung erlangen.
Die Diagnose einer AML erfordert:
- den Nachweis eines Anteils unreifer Blasten von mindestens 30 % (FAB-Klassifikation) bzw. 20 % (WHO-Klassifikation) im Knochenmark.
- die Zuordnung der Blasten zur myeloischen Reihe durch zytochemische Untersuchung und/oder Immunphänotyp.
- die weitere Zuordnung zu einem AML-Subtyp entsprechend der FAB-Klassifikation oder WHO-Klassifikation.
Morphologie und Zytochemie
Grundlage der Diagnostik ist die mikroskopische Untersuchung von Knochenmarkausstrichen. Charakteristische Merkmale wie z. B. der Nachweis von Auerstäbchen ermöglichen die Zuordnung der Blasten zur myeloischen Reihe. Bei Auerstäbchen handelt es sich um feine, stäbchenförmige Granula oder große, ovale bis elliptiforme Einschlüsse (Auer-Körper) im Zytoplasma unreifer Leukämiezellen. Ggf. mit Hilfe zusätzlicher zytochemischer Untersuchungen (Peroxidase, Esterase, PAS-Reaktion) gelingt in der Mehrzahl der Fälle die Abgrenzung einer AML von einer ALL und die Einordnung entsprechend der FAB-Klassifikation (French-American-British).
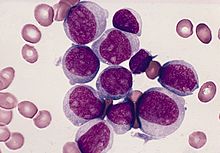 Blasten bei AML FAB M1
Blasten bei AML FAB M1
FAB-Subtyp Bezeichnung Zytogenetische Aberrationen M0 Akute myeloische Leukämie mit minimaler Differenzierung - M1 Akute myeloische Leukämie ohne Ausreifung - M2 Akute myeloische Leukämie mit Ausreifung t(8;21) M3 Akute Promyelozyten-Leukämie (APL) t(15;17) M3v Akute Promyelozyten-Leukämie, mikrogranuläre Form t(15;17) M4 Akute myelomonozytäre Leukämie - M4Eo Akute myelomonozytäre Leukämie mit Eosinophilie inv(16) M5a Akute Monoblasten-Leukämie - M5b Akute Monozyten-Leukämie - M6 Akute Erythroleukämie - M7 Akute Megakaryoblasten-Leukämie - Eine Weiterentwicklung der FAB-Klassifikation stellt die WHO-Klassifikation dar, die häufige chromosomale Aberrationen bei der AML mit einbezieht. Die Promyelozytenleukämie (FAB M3 bzw. M3v) weist klinische, biologische und auch therapeutische Besonderheiten auf und wird in einem eigenen Artikel besprochen.
Immunphänotypisierung
Die Immunphänotypisierung hat bei der AML vorwiegend bestätigenden Charakter, kann aber in Zweifelsfällen wichtige zusätzliche Informationen liefern. Mit Hilfe von markierten monoklonalen Antikörpern wird die Expression membranständiger Oberflächenmoleküle auf den Leukämiezellen untersucht. In den meisten Fällen handelt es sich um Differenzierungs-Antigene, die auch im Verlauf der normalen Hämatopoese exprimiert werden und in der sog. CD-Nomenklatur zusammengefasst werden. Die für die Charakterisierung einer AML wichtigsten Antigene sind in folgender Tabelle zusammengefasst.
Antigen Pan-myeloisch CD13, CD33, CD65s Granulozytär, monozytär CD15, CD14, CD64 Vorläufer-Zellen CD34, CD117, CD7 Zytogenetik
Bei den meisten Patienten mit AML lassen sich erworbene numerische und strukturelle chromosomale Aberrationen in den Leukämiezellen nachweisen. Einige dieser Aberrationen sind eng mit einem bestimmten morphologischen und klinischen Subtyp verbunden bzw. definieren diesen. In den letzten Jahren ist die diagnostische, prognostische und therapeutische Bedeutung der zytogenetischen Veränderungen bei AML zunehmend klar geworden. Häufige chromosomale Aberrationen sind:
Aberration Besondere Merkmale +8 Häufigste numerische Chromosomenaberration bei AML t(8;21) Häufigste strukturelle Chromosomenaberration bei AML t(15;17) Spezifisch für Promyelozytenleukämie t(9;22) sog. „Philadelphia-Chromosom“ inv(16) M4Eo Therapie und Prognose
Prognostische Faktoren
Man unterscheidet eine de novo-AML von einer sekundären AML. Beide Formen unterscheiden sich hinsichtlich der Biologie der Erkrankung und dem therapeutischen Ansprechen. Von einer sekundären AML spricht man, wenn der Erkrankung eine hämatologische Erkrankung, z. B. ein myelodysplastisches Syndrom, vorangegangen ist oder eine Exposition gegenüber Zytostatika oder Strahlen bestand. Die sekundäre AML weist einige prognostisch ungünstige zytogenetische Veränderungen auf und spricht auf die Therapie schlechter an.
Anhand einer Reihe von prognostischen Faktoren lässt sich das Ansprechen auf eine Therapie abschätzen. Diese Faktoren sind allerdings nicht voneinander unabhängig, z. B. sind komplexe zytogenetische Aberrationen bei sekundärer AML häufiger.
Prognostisch ungünstig sind u. a.:
- Hohes Alter und/oder schlechter Allgemeinzustand bei Diagnosestellung
- Hohe Leukozytenzahl
- Vorangegangene hämatologische Erkrankung (z. B. MDS, Blastenschub bei CML), sekundäre AML
- FAB M0, FAB M6, FAB M7
- Ungünstige Zytogenetik (Trisomie 8, Aberrationen von Chromosom 5 oder 7, t(6;9), t(9;22), 11q23-Aberrationen, komplexe zytogenetische Aberrationen)
- Sekundäre Leukämie
Prognostisch günstige Faktoren sind:
- Promyelozyten-Leukämie (FAB M3)
- Günstige Zytogenetik: t(15;17), inv(16), t(8;21), normaler Karyotyp
Therapiedurchführung
Im Dezember des Jahres 2008 hat die Europäische Kommission Azacitidin (Vidaza®) als Orphan Drug zur Therapie myelodysplastischer Syndrome (MDS) zugelassen. Die Indikation umfasst die Behandlung von Hochrisiko-MDS-Patienten der Kategorien »Int-2« und »High-Risk« nach dem International Prognostic Scoring System (IPSS). Weitere indizierte Subgruppen nach der WHO-Klassifikation sind die chronisch myelomonozytäre Leukämie (CMML) mit 10 bis 29 Prozent Blasten im Knochenmark ohne myeloproliferative Störung sowie die akute myeloische Leukämie (AML) mit 20 bis 30 Prozent Blasten im Knochenmark und gleichzeitiger Mehrlinien-Dysplasie (RAEB-t nach FAB-Klassifikation).[1][2]
Grundlage der Therapie der AML ist eine intensive Chemotherapie. Ziel der sog. Induktionstherapie ist das Erreichen einer kompletten Remission, d. h. einer Beseitigung aller Krankheitssymptome mit Normalisierung des Blutbildes und Beseitigung der pathologischen Zellpopulation im Knochenmark (Blasten < 5 %). Die Behandlung besteht aus mehrtägigen Therapieblöcken, die mehrfach (zwei bis drei Mal) wiederholt werden. Bei diesen kommt der Patient jeweils in sogenannte „Isolation“; dabei ist die Zahl der Leukozyten so niedrig, dass jeder Infekt unter Umständen tödlich sein kann, was absolute Mundschutzpflicht beim Patientenumgang zur Folge hat. Wichtige Medikamente sind Cytarabin und die Anthracycline Daunorubicin bzw. Idarubicin. Gegebenenfalls wird zusätzlich Thioguanin gegeben. Ist eine komplette Remission erreicht (meist nach 1–2 Therapieblöcken), folgt die Postremissionstherapie. Sie umfasst die Konsolidierungstherapie (hochdosiertes Cytarabin) und bei der Akuten Promyelozytenleukämie eine Erhaltungstherapie (All-trans Retinol, 6-Mercaptopurin und Methotrexat). Als weitere Therapieverfahren kommen die allogene oder autologe Stammzelltransplantation in Betracht.
Der Stellenwert neuer Therapieansätze, wie beispielsweise das Immuntoxin Gemtuzumab-Ozogamicin, das nur in den Vereinigten Staaten zugelassen ist, ist bisher noch unklar.
In der Begleittherapie spielt der Ersatz von Blutbestandteilen (Erythrozyten, Thrombozyten), evtl. der Einsatz von Wachstumsfaktoren für Granulozyten (z. B. G-CSF) und die Bekämpfung von Infektionen mittels Antibiotika gegen bakterielle Infektionen und Antimykotika gegen Pilzinfektionen eine wesentliche Rolle.
Therapieergebnisse
Mit der Induktionstherapie gelingt es, je nach Patientenauswahl, bei etwa 70% der Patienten mit AML eine komplette Remission zu erreichen.
Die Therapieergebnisse bei älteren Patienten (> 60 Jahre) sind deutlich schlechter, die Rate kompletter Remissionen liegt hier zwischen 30 % und 60 %. Ursache sind nicht nur Begleiterkrankungen im höheren Lebensalter, die zu Komplikationen führen, sondern auch das vermehrte Auftreten prognostisch ungünstiger Faktoren wie ungünstige Zytogenetik oder Sekundärleukämien nach vorangegangenem MDS. Leider ist bei der Mehrheit der Patienten mit einem Rückfall zu rechnen, etwa 15 %–25 % erreichen Langzeitremissionen nach konventioneller Chemotherapie und können als geheilt angesehen werden. Günstiger sind die Ergebnisse nach allogener Stammzelltransplantation.
Wesentlich bessere Therapieresultate ergeben sich für die Promyelozytenleukämie.
Forschung
Die Deutsche Krebshilfe, die regelmäßig hohe Finanzmittel aus Spendengeldern der Krebsforschung zuführt, hat 2010 rund 300.000 Euro allein für eine Studie zur Erforschung der Ursachen von Leukämie bereitgestellt. Die Wissenschaftler um Dr. Philipp A. Greif und Prof. Stefan K. Bohlander von der Medizinischen Klinik und Poliklinik III der Ludwig-Maximilians-Universität München verfolgen nach Angaben der von der Ärztin Mildred Scheel gegründeten Hilfsorganisation eine neue Strategie: Sie wollen die an der Entstehung von Leukämie beteiligten Gene identifizieren.
Das Verständnis der genetischen Mechanismen, die zu einer AML führen, soll zukünftig die Entwicklung neuer Behandlungsmethoden ermöglichen, erklärte der Hauptgeschäftsführer der Deutschen Krebshilfe, Gerd Nettekoven. Ziel des Projektes ist es, möglichst viele solcher Gen-Mutationen zu identifizieren, betonen die Wissenschaftler. „Unsere Arbeit ist wegbereitend, um in Zukunft die individuellen genetischen Ursachen einer Krebserkrankung zu erkennen. Damit schaffen wir die Voraussetzung für die Entwicklung neuer und zielgerichteter Behandlungsmethoden“, sagte Dr. Greif.
Literatur
Allgemein
- Löwenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med 1999; 341:1051–62. Review. PMID 10502596
Diagnostik
- Bennett, JM, Catovsky, D, Daniel, MT, et al. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann Intern Med 1985; 103:620–5. PMID 3862359
- Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 2002; 100:2292–302. PMID 12239137
Weblinks
- Leitlinie der „Deutschen Gesellschaft für Hämatologie und Onkologie“ zu den Akuten Leukämien (PDF-Datei) (141 kB)
- MDS-Krankheit.de Umfangreiche Informationen für Patienten
- Vidaza.de Umfangreiche Informationen für Fachkreise und Patienten
- Audio-Beitrag zu akuten Leukämien
- Uni Ulm: Akute Myeloische Leukämie: Gensignatur gibt Hinweise auf Krankheitsverlauf. (Pressemitteilung bei idw)
- http://www.kinderkrebsinfo.de/AML
Einzelnachweise
- ↑ Europäischer öffentlicher Beurteilungsbericht (EPAR), European Medicines Agency (EMA)
- ↑ Azacitidin in der Europäischen Union zur Therapie myelodysplastischer Syndrome zugelassen, Journal Onkologie 2009

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Kinderonkologie
- Leukämie

Wikimedia Foundation.