- Ganglioneurom
-
Klassifikation nach ICD-10 D35 Gutartige Neubildung sonstiger und nicht näher bezeichneter endokriner Drüsen D35.0 Nebenniere D36 Gutartige Neubildung an sonstigen und nicht näher bezeichneten Lokalisationen D36.1 Periphere Nerven und autonomes Nervensystem ICD-10 online (WHO-Version 2011) Klassifikation nach ICD-O-3 9490/0 Ganglioneurom ICD-O-3 online 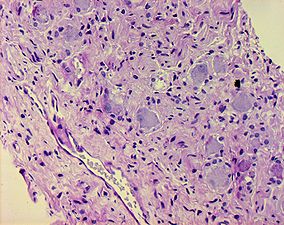 Histologie des Ganglioneuroms mit reifen Ganglienzellen und spindelförmigen Schwann-Zellen.
Histologie des Ganglioneuroms mit reifen Ganglienzellen und spindelförmigen Schwann-Zellen.
Ganglioneurome sind seltene, im allgemeinen gutartige Tumoren des sympathischen Nervensystems, welche sich feingeweblich aus reifen Ganglienzellen, Schwann-Zellen, Bindegewebe und Nervenfasern zusammensetzen. Sie können prinzipiell überall dort entstehen, wo sich sympathisches Nervengewebe findet, insbesondere im Nebennierenmark, den sympathischen Ganglien beidseits der Wirbelsäule, dem hinteren Mediastinum, Kopf oder Hals. Seltene Lokalisationen umfassen die Harnblase, die Darm- oder Bauchwandung sowie die Gallenblase. Der Tumor entsteht entweder sporadisch oder durch Ausreifung aus den unreiferen Ganglioneuroblastomen oder Neuroblastomen[1]. Selten soll eine Assoziation des Tumors mit syndromalen Erkrankungen wie der Neurofibromatose Typ 1, dem Beckwith-Wiedemann-Syndrom, dem Morbus Hirschsprung oder dem DiGeorge-Syndrom bestehen. Knapp 40 % der Ganglioneurome produzieren Katecholamine[2]. Während es sich bei Ganglioneuromen um Tumoren des peripheren Nervensystems handelt, ist die Bezeichnung Ganglioneurom des zentralen Nervensystems eine obsolete Bezeichnung für einen seltenen gutartigen hirneigenen Tumor, das Gangliozytom.
Inhaltsverzeichnis
Epidemiologie
Wenngleich sich Ganglioneurome in jedem Lebensalter manifestieren können, sind in der Mehrzahl der Fälle Kinder, Jugendliche und junge Erwachsene betroffen. Das mittlere Erkrankungsalter liegt bei 7 Jahren. In Einzelfällen wurden auch angeborene Ganglioneurome beschrieben[3]. Der Tumor zeigt keine deutliche Geschlechtsbevorzugung.
Morphologie
Ganglioneurome treten häufig als gut umschriebene, rundliche oder gelappte Tumoren von fester Konsistenz in Erscheinung. Die Schnittfläche erscheint grau-weiß bis gelb-grau und kann zystische oder verfettete Areale sowie Verkalkungen zeigen. Histologisch finden sich spindelige Zellproliferate (Schwann-Zellen) ähnlich wie bei einem Neurofibrom, wobei jedoch zusätzlich an Ganglienzellen erinnerende ganglioide Tumorzellen, häufig in Gruppen gelagert und mehrkernig, zu erkennen sind. Fokal werden häufig lymphozytäre Infiltrate sichtbar. Nekrosen fehlen üblicherweise[4].
Klinische Symptomatik
Unabhängig von ihrer Größe sind Ganglioneurome häufig asymptomatisch und werden nicht selten zufällig im Rahmen von Routineuntersuchungen entdeckt. Mögliche Symptome sind Bauchschmerzen, Kurzatmigkeit, Husten oder eine tastbare Resistenz im Bauchraum. Hormonproduzierende Ganglioneurome können zu Bluthochdruck, Hautrötung oder Vermännlichung führen. Wässrige Durchfälle können aus einer Sekretion von vasoaktivem intestinalem Polypeptid (VIP) durch den Tumor resultieren[5].
Diagnose
Zur Diagnose eignen sich bildgebende Verfahren wie die Magnetresonanztomographie (MRT), Computertomographie (CT) oder die MIBG-Szintigrafie. Bei letzterem Verfahren macht man sich die Tatsache zunutze, dass zumindest ein Teil der Ganglioneurome den Stoff MIBG aufnimmt und innerhalb der Tumorzellen anreichert. Eine Markierung durch ein radioaktives Jod-Isotop gewährleistet in diesen Fällen die Detektierbarkeit mittels Szintigraphie. Die MIBG-anreichernden Tumoren sind diejenigen Ganglioneurome, die gleichzeitig auch Katecholamine produzieren. Eine Biopsie oder komplette Tumorentfernung mit anschließender histologischer Untersuchung des gewonnenen Gewebes kann zur definitiven Diagnosesicherung erforderlich sein. Im Falle einer Hormonproduktion durch den Tumor sind laborchemische Untersuchungen sinnvoll.
Therapie und Prognose
Therapie der Wahl ist in der Regel die chirurgische Entfernung des Tumors. Nur selten werden Rückfälle beobachtet, so dass üblicherweise von einer exzellenten Prognose auszugehen ist. Eine Metastasierung von Ganglioneuromen[6][7] wird ebenso wie die maligne Transformation in ein Ganglioneuroblastom oder Neuroblastom[8] in Einzelfällen beschrieben. Diskutiert wird die Möglichkeit, dass ganglioneuromtypisch differenzierte Metastasen auch durch gewebliche Ausreifung aus Absiedlungen von Neuroblastomen und Ganglioneuroblastomen entstehen könnten, so dass Ganglioneurome selbst möglicherweise kein metastatisches Potenzial besitzen.
Differentialdiagnose
Bei der Diagnose auszuschließen sind andere Tumoren von Nebenniere bzw. sympathischem Nervensystem, wie das Nebennierenadenom und -karzinom, das Neuroblastom und das Ganglioneuroblastom, neuroblastische Mischtumoren oder das Phäochromozytom.
Literatur
Jedynak, A.R. et al.: Ganglioneuroma and Ganglioneuroblastoma, 2008; http://www.emedicine.com/radio/topic293.htm
Einzelnachweise
- ↑ Dyke, C. et al.: Maturation of ganglioneuroblastoma to ganglioneuroma. Cancer 2006; 20(8): 1343-1349.
- ↑ Geoerger, B. et al. Metabolic activity and clinical features of primary ganglioneuromas. Cancer 2001; 91(10): 1905-1913.
- ↑ Ponce-Camacho M.A. et al.: A 5-year-old girl with a congenital ganglioneuroma diagnosed by fine needle aspiration biopsy : A case report. CytoJournal 2008;5:5
- ↑ PathologyOutlines.com, Mediastinum (4. November 2007), http://www.pathologyoutlines.com/mediastinum.html#ganglioneuroma
- ↑ Swift P.G. et al.: Watery diarrhoea and ganglioneuroma with secretion of vasoactive intestinal peptide. Archives of Disease in Childhood 1975; 50: 896-899.
- ↑ Garvin, J.H. et al.: Ganglioneuroma presenting with differentiated skeletal metastases. Report of a case. Cancer 2006; 54(2), 357-360.
- ↑ Goya A. et al.: Mediastinal ganglioneuroma with lymph node metastasis in a boy presenting upper airway infection. Journal of Chinese Clinical Medicine 2006; 1(6): 326-329.
- ↑ Kulkani, A.V. et al.: Malignant transformation of ganglioneuroma into spinal neuroblastoma in an adult. Case report. Journal of Neurosurgery 1998; 88(2):324-327.

Bitte den Hinweis zu Gesundheitsthemen beachten!
Wikimedia Foundation.