- Holstein-Herring-Methode
-
Die Holstein-Herring-Methode[1][2][3][4], auch bekannt unter der englischen Bezeichnungen surface integral method[5][6] oder Smirnov's method[7], ist ein effektives Verfahren zur Berechnung der Austauschenergieaufspaltung asymptotisch entarteter Energiezustände in molekularen Systemen. Obwohl die Austauschenergieaufspaltung für zunehmend größer werdende internukleare Abstände R immer schwieriger zu berechnen ist, hat sie fundamentale Bedeutung für die Theorien der Bindung in Molekülen und des Magnetismus.
Inhaltsverzeichnis
Theorie
Die Grundidee der Holstein-Herring-Methode lässt sich am Beispiel des Wasserstoffmolekülions, oder allgemeiner, der Atom-Ion-Systeme oder „Systeme mit einem aktiven Elektron“ folgendermaßen illustrieren. Wir betrachten Molekülzustände, die durch Zustandsfunktionen beschrieben werden, welche sich unter Rauminversion gerade oder ungerade verhalten. Dies wird durch die Suffixe g und u gekennzeichnet und ist Standard zur Kennzeichnung elektronischer Zustände zweiatomiger Moleküle (für Atomzustände sind dagegen die englischen Ausdrücke „even“ und „odd“ gebräuchlich). Die zugehörige elektronische Schrödinger-Gleichung lässt sich schreiben als:
wobei E die (elektronische) Energie eines gewählten quantenmechanischen Zustands (Eigenzustands) ist, mit einer elektronischen Zustandsfunktion
 die von den Ortskoordinaten des Elektrons abhängt, und wobei V das Coulomb-Potential der Elektron-Kern-Wechselwirkung ist. Für das Wasserstoff-Molekülion gilt:
die von den Ortskoordinaten des Elektrons abhängt, und wobei V das Coulomb-Potential der Elektron-Kern-Wechselwirkung ist. Für das Wasserstoff-Molekülion gilt:Für irgendeinen geraden Zustand lässt sich die elektronische Schrödinger-Gleichung in atomaren Einheiten (
 ) schreiben als:
) schreiben als:Für irgendeinen ungeraden Zustand lässt sich die zugehörige Wellengleichung schreiben als:
Der Einfachheit halber nehmen wir reelle Funktionen an (obwohl das Endergebnis für den Fall komplexer Funktionen verallgemeinert werden kann). Nun multiplizieren wir die Gleichung für die gerade Funktion von links mit ψ − , die Gleichung für die ungerade Funktion von links mit ψ + , und erhalten daraus die Differenz:
wobei ΔE = E − − E + die Austauschenergieaufspaltung ist. Im nächsten Schritt definieren wir, ohne Beschränkung der Allgemeinheit, orthogonale Ein-Teilchen-Funktionen,
 und
und  , die an den Kernen lokalisiert seien und schreiben:
, die an den Kernen lokalisiert seien und schreiben:Dies ist ähnlich dem in der Quantenchemie verwendeten LCAO-Ansatz (Linear combination of atomic orbitals molecular orbital method), wir müssen aber betonen, dass die Funktionen
und im allgemeinen „polarisiert“ sind, d.h. sie sind keine reinen Eigenfunktionen der Drehimpulsoperatoren bzgl. ihrer jeweiligen Zentren (s.a. unten). Allerdings reduzieren sich die lokalisierten Funktionen  im Grenzfall
im Grenzfall  auf die wohlbekannten atomaren (wasserstoff-artigen) Psi-Funktionen
auf die wohlbekannten atomaren (wasserstoff-artigen) Psi-Funktionen  . Wir bezeichnen nun mit M die Ebene senkrecht zur Kernverbindungslinie in der Mitte zwischen beiden Kernen (s. Diagram für Wasserstoff-Molekülion für weitere Einzelheiten), mit
. Wir bezeichnen nun mit M die Ebene senkrecht zur Kernverbindungslinie in der Mitte zwischen beiden Kernen (s. Diagram für Wasserstoff-Molekülion für weitere Einzelheiten), mit  einen Einheitsvektor senkrecht zu dieser Ebene (dieser Vektor sei parallel zur kartesischen z-Richtung), so dass der gesamt dreidimensionale Raum
einen Einheitsvektor senkrecht zu dieser Ebene (dieser Vektor sei parallel zur kartesischen z-Richtung), so dass der gesamt dreidimensionale Raum  in einen linken (L) und einen rechten (R) Halbraum geteilt wird. Aus Symmetrieüberlegungen folgt:
in einen linken (L) und einen rechten (R) Halbraum geteilt wird. Aus Symmetrieüberlegungen folgt:Dies impliziert, dass:
Die lokalisierten Funktionen sind normiert, so dass gelten muss:
und umgekehrt. Integration dieses Ergebnisses über den gesamten Raum links der Ebene M ergibt:
und
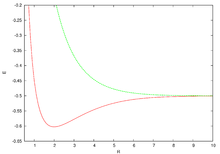 Energie (E) der beiden niedrigsten gebundenen Zustände des Wasserstoff-Molekülions
Energie (E) der beiden niedrigsten gebundenen Zustände des Wasserstoff-Molekülions
 , als Funktion des Kern-Kern-Abstandes (R) in atomaren Einheiten.
, als Funktion des Kern-Kern-Abstandes (R) in atomaren Einheiten.Anwendung einer Variante des gaußschen Integralsatzes auf dieses Ergebnis führt schließlich auf die Holstein-Herring-Formel:
wobei
 ein differentielles Flächenelement der Mittelebene M ist. Mit dieser Formel gelang es Herring erstmals zu zeigen,[3] dass der führende Term der asymptotischen Entwicklung der Energiedifferenz zwischen den beiden niedrigsten Zuständen des Wasserstoff-Molekülions, also des ersten angeregten Zustandes 2pσu und des Grundzustandes 1sσg (bezeichnet nach molekularer Notation – s. obige Abbildung für die Energiekurven), folgende mathematische Form hat:
ein differentielles Flächenelement der Mittelebene M ist. Mit dieser Formel gelang es Herring erstmals zu zeigen,[3] dass der führende Term der asymptotischen Entwicklung der Energiedifferenz zwischen den beiden niedrigsten Zuständen des Wasserstoff-Molekülions, also des ersten angeregten Zustandes 2pσu und des Grundzustandes 1sσg (bezeichnet nach molekularer Notation – s. obige Abbildung für die Energiekurven), folgende mathematische Form hat:Vorherige Berechnungen auf der Basis der LCAO-Näherung für die atomaren Orbitale hatten fälschlicherweise den Vorfaktor 4 / 3 anstatt 4 / e ergeben.
Anwendungen
Die Holstein-Herring-Formel hatte nur begrenzte Bedeutung für Anwendungen, bis um 1990, als Tang, Toennies, und Yiu[8] zeigten, dass
eine polarisierte Funktion sein kann, d. h. eine atomare, an einem der beiden Kernorte lokalisierte Wellenfunktion, die durch den Einfluss des anderen Kerns verzerrt wird und daher keine eindeutige Symmetrie (gerade oder ungerade) mehr aufweist. Dennoch kann die oben angegebene Holstein-Herring-Formel verwendet werden, und liefert die korrekte asymptotische Reihenentwicklung für die Austauschenergieaufspaltung. Auf diese Weise ist auch ein ursprüngliches Zwei-Zentren-Problem erfolgreich in ein effektives Ein-Zentren-Problem umgewandelt worden. Anschließend wurde diese Formel für Zwei-Zentren-Probleme mit einem aktiven Elektronen (z. B. Alkalidimer-Kationen) erweitert. Durch Scott et al. wurde das Verständnis dieses zunächst überraschenden Ergebnisses vertieft, was die Klärung subtiler, aber wichtiger Fragen zur Konvergenz der polarisierten Funktionen erforderte[9][10][11]. Das Ergebnis dieser Analyse bedeutet, dass im Prinzip jede beliebige Ordnung der asymptotischen Reihenentwicklung der Austauschenergieaufspaltung berechnet werden kann. Die Holstein-Herring-Methode ist auch für den Fall von zwei aktiven Elektronen erweitert worden, d. h. für die beiden niedrigsten gebundenen Zustände des Wasserstoff-Moleküls H2[12] und allgemeinere zweiatomige Systeme[13].Physikalische Interpretation
Die oben angegebene Holstein-Herring-Formel kann wie folgt physikalisch interpretiert werden: Das Elektron tunnelt zwischen beiden Kernen hin und her, erzeugt dadurch einen Strom, dessen Flussdichte durch die Mittelebene M die Bestimmung der Austauschenergieaufspaltung erlaubt. Bezogen auf den Tunneleffekt, eine ergänzende Auslegung von Sidney Coleman's "Aspects of Symmetry" ("Aspekte der Symmetrie", 1985) hat eine "Instanton" Reise in die Nähe und über den klassischen Weg innerhalb Pfadintegral. Diese Energie wird also von beiden Kernen geteilt, d. h. ausgetauscht. Zu beachten ist noch, dass das Volumenintegral über R im Nenner der Holstein-Herring-Formel subdominant ist, so dass für genügend große Kern-Kern-Abstände der Nenner einfach gleich eins gesetzt werden kann und nur das Oberflächenintegral im Zähler berechnet zu werden braucht.
Einzelnachweise
- ↑ Holstein T., „Mobilities of positive ions in their parent gases“, J. Phys. Chem. 56, 832-836 (1952).
- ↑ Holstein T., Westinghouse Research Report 60-94698-3-R9, (unpublished), (1955).
- ↑ a b Herring C., „Critique of the Heitler-London Method of Calculating Spin Couplings at Large Distances“, Rev. Mod. Phys. 34, 631-645 (1962).
- ↑ Bardsley J.N., Holstein T., Junker B.R., and Sinha S., „Calculations of ion-atom interactions relating to resonant charge-transfer collisions“, Phys. Rev. A 11, 1911-1920 (1975).
- ↑ T.C. Scott, M. Aubert-Frécon, D. Andrae, „Asymptotics of Quantum Mechanical Atom-Ion Systems“, AAECC (Applicable Algebra in Engineering, Communication and Computing) 13, 233-255 (2002).
- ↑ Aubert- Frécon M., Scott T.C., Hadinger G., Andrae D., Grotendorst J., and Morgan III J.D., „Asymptotically Exact Calculation of the Exchange Energies of One-Active-Electron Diatomic Ions with the Surface Integral Method“, J. Phys. B: At. Mol. Opt. Phys., 37, pp. 4451-4469 (2004).[1]
- ↑ Smirnov B.M. and Chibisov M.I., „Electron exchange and changes in the hyperfine state of colliding alkaline metal atoms“ Sov. Phys. JETP 21, 624-628 (1965).
- ↑ Tang K.T., Toennies J.P., and Yiu C.L., „The exchange energy of H2+ calculated from polarization perturbation theory“, J. Chem. Phys. 94, 7266-7277 (1991).
- ↑ Scott T.C., Dalgarno A. and Morgan III J.D. (1991). „Exchange Energy of H2+ Calculated from Polarization Perturbation Theory and the Holstein-Herring Method“, Phys. Rev. Lett. 67: 1419-1422.[2]
- ↑ Scott T.C., Babb J.F., Dalgarno A. and Morgan III J.D.,„Resolution of a Paradox in the Calculation of Exchange Forces for H2+, Chem. Phys. Lett. 203, 175-183 (1993).[3]
- ↑ Scott T.C., Babb J.F., Dalgarno A. and Morgan III J.D., „The Calculation of Exchange Forces: General Results and Specific Models“, J. Chem. Phys., 99,2841-2854, (1993). [4]
- ↑ Herring C., and Flicker M.,„Asymptotic Exchange Coupling of Two Hydrogen Atoms“, Phys. Rev. A 134, 362-366 (1964).
- ↑ Scott T.C., Aubert-Frécon M., Andrae D., Grotendorst J., Morgan III J.D. and Glasser M.L., „Exchange Energy for Two-Active-Electron Diatomic Systems Within the Surface Integral Method“, AAECC, 15, 101-128 (2004). doi:10.1007/s00200-004-0156-6













Wikimedia Foundation.