- Common Variable Immunodeficiency
-
Klassifikation nach ICD-10 D83 Variabler Immundefekt (common variable immunodeficiency) D83.0 Variabler Immundefekt mit überwiegenden Abweichungen der B-Zellen-Zahl und -Funktion D83.1 Variabler Immundefekt mit überwiegenden immunregulatorischen T-Zell-Störungen D83.2 Variabler Immundefekt mit Autoantikörpern gegen B- oder T-Zellen D83.8 Sonstige variable Immundefekte D83.9 Variabler Immundefekt, nicht näher bezeichnet ICD-10 online (WHO-Version 2006) Das variable Immundefektsyndrom (engl. Common Variable Immunodeficiency; CVID) ist der beim Menschen am häufigsten zu einer symptomatischen Erkrankung führende angeborene Immundefekt. Er ist vor allem gekennzeichnet durch den Mangel an einer bestimmten Klasse von Antikörpern, dem Immunglobulin G. Die genaue Ursache der Erkrankung ist unbekannt, es handelt sich aber mit großer Wahrscheinlichkeit um eine heterogene Gruppe verschiedener einzelner Krankheiten. Betroffene Patienten leiden meist an einer Häufung von Atemwegsinfektionen, daneben gibt es eine Vielzahl weiterer möglicher Symptome. Neben Infektionen verschiedener Körperteile (vor allem durch Bakterien) gehören dazu auch Autoimmunphänomene und Krebserkrankungen. Üblicherweise wird die Erkrankung mit Infusionen von Immunglobulin G behandelt.
Inhaltsverzeichnis
Ursachen
Nur bei den wenigsten Fällen des variablen Immundefektsyndroms lässt sich die genaue Ursache nach derzeitigem Stand der Forschung ermitteln. Bei einigen Betroffenen hat man Mutationen im TNFRSF13B-Gen auf Chromosom 17 Genlocus p11.2 gefunden, die ursächlich mit dem variablen Immundefektsyndrom in Zusammenhang gebracht werden[1]. Es treten sowohl familiäre wie sporadische Formen der Erkrankung auf.
Insgesamt ist anzunehmen, dass es sich beim variablen Immundefektsyndrom um eine heterogene Gruppe unterschiedlicher Einzelkrankheiten mit unterschiedlichen Ursachen handelt, die bisher zum größten Teil nicht bekannt sind.
Häufigkeit und Vorkommen (Epidemiologie)
Die Häufigkeit des variablen Immundefektsyndroms wird in Deutschland auf 1 pro 25.000 Personen geschätzt, die Schätzungen für Industrienationen liegen dabei zwischen 1:10.000 und 1:100.000. Man nimmt an, dass in Deutschland zwischen 800 und 3.200 Menschen betroffen sind. Es handelt sich also insgesamt um eine seltene Krankheit, auch wenn es einer der häufigsten angeborenen Immundefekte ist.
Bei der Altersverteilung zum Zeitpunkt der Diagnosestellung gibt es zwei „Gipfel“: Die frühe Form wird meist im Kleinkindalter festgestellt, die späte Form im jungen Erwachsenenalter.
Klinik und Symptome
Die Symptome des variablen Immundefektsyndroms kann man folgenden Gruppen zuordnen: Infektionen, Störungen des Magen-Darm-Trakts, chronische Atemwegserkrankungen, Hauterscheinungen, Veränderungen lymphatischer Gewebe, Granulome, Autoimmunphänomene und Tumoren.
Infektionen:
- Atemwegsinfektionen durch bekapselte Bakterien (z. B. Haemophilus influenzae, Streptococcus pneumoniae, Moraxella catharralis)
- Entzündungen des Gehirns (Enzephalitiden) durch Enteroviren
- chronische Durchfallerkrankung durch Lamblien
- Infektionen des Harntrakts durch Mykoplasmen
Störungen des Magen-Darm-Trakts:
- Durchfall (ca. 1/3 der Patienten)
- ungenügende Aufnahme von Nährstoffen (Malabsorption)
Chronische Atemwegserkrankungen:
- Höhlenartige Erweiterungen der unteren Atemwege (Bronchiektasie)
Veränderungen lymphatischer Gewebe:
- Vergrößerung von Leber und Milz (Hepato-Splenomegalie)
Granulome:
- Entzündungsherde mit bestimmtem Aufbau in inneren Organen wie Leber, Milz, Lunge und Knochenmark
Autoimmunphänomene:
- reaktive Gelenkentzündungen (reaktive Arthritiden)
- immunologisch bedingter Mangel an Blutplättchen (Immunthrombozytopenie, ca. 20 % der Patienten)
- immunologisch bedingte Blutarmut (autoimmunhämolytische Anämie)
- perniziöse Anämie (ca. 10 % der Patienten)
Hauterscheinungen:
- Haarausfall (teilweise als Alopecia areata)
- Weißfleckenkrankheit (Vitiligo)
- Granulome der Haut
Diagnose
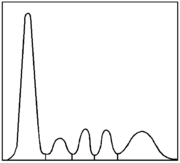 Serumelektrophorese eines Gesunden (schematisch)
Serumelektrophorese eines Gesunden (schematisch)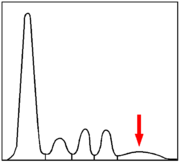 Serumelektrophorese eines CVID-Patienten (schematisch)
Serumelektrophorese eines CVID-Patienten (schematisch)Der Verdacht auf ein variables Immundefektsyndrom ergibt sich häufig aus immer wiederkehrenden Infektionen der Atemwege. Es kann sich aber auch um einen Zufallsbefund handeln, der durch eine Serumelektrophorese oder eine Bestimmung der Immunglobuline aus anderen Gründen heraus entsteht. In aller Regel fällt bei betroffenen Patienten bereits in der Serumelektrophorese eine Verminderung der Gamma-Globulin-Fraktion auf.
Zunächst wird die Verdachtsdiagnose durch quantitative Bestimmung der Immunglobuline gestellt. Das Immunglobulin G im Serum ist immer erniedrigt, in der Regel liegt es unter 3 g/l. Häufig sind die Immunglobuline A und M ebenfalls vermindert. Ein wesentlicher Baustein der Diagnose ist also ein Antikörpermangel im Blut.
Um die Diagnose eines variablen Immundefektsyndroms definitiv stellen zu können, müssen andere mögliche Ursachen für den Antikörpermangel ausgeschlossen werden. Dazu gehören z. B. eine monoklonale Vermehrung von Immunglobulin-Leichtketten (Bence-Jones-Myelom), ein starker Eiweißverlust über die Niere (nephrotisches Syndrom) oder den Darm (exsudative Enteropathie).
Außerdem sind einige immunologische Spezialuntersuchungen wie die Bestimmung der Immunglobulin-G-Subklassen, die durchflusszytometrische Analyse der peripheren Blutlymphozyten und die Messung der spezifischen Antikörperproduktion notwendig.
Differentialdiagnose
- Agammaglobulinämie Bruton (XLA)
- autosomal-rezessiv vererbte Agammaglobulinämie (Schweizer Typ)
- selektiver Immunglobulin-A-Mangel
- Hyper-IgM-Syndrom
- transitorische Hypogammaglobulinämie im Kindesalter
- Kombinierte Immundefekte (SCID etc.)
- multiples Myelom (insbesondere Bence-Jones-Myelom)
- nephrotisches Syndrom (Eiweißverlust über die Nieren)
- exsudative Enteropathie (Eiweißverlust über den Darm)
Therapie
Eine Behandlung ist nur bei Patienten notwendig, die durch die Erkrankung an Beschwerden leiden. Eine vorbeugende Behandlung bei beschwerdefreien Patienten ist nicht angezeigt. Als wirksame Behandlungsmöglichkeit steht die regelmäßige intravenöse Infusion von Immunglobulinen (abgekürzt IVIG) zur Verfügung. Die Infusionen werden alle 2–6 Wochen in einer Dosierung von 200 bis 600 mg pro kg Körpergewicht verabreicht. Ziel dieser Behandlung ist es, das Immunglobulin G im Serum stets oberhalb von 5 g/l zu halten. Zusätzlich werden alle im Rahmen der Erkrankung auftretenden bakteriellen Infektionen mit Antibiotika behandelt.
Prognose
Insgesamt ist nicht zu erwarten, dass Patienten mit variablem Immundefektsyndrom eine ähnliche Lebenserwartung haben wie Gesunde. Die Datenlage dazu ist allerdings spärlich. Als Faktoren, die die Prognose der Erkrankung begrenzen, werden in der Literatur vor allem genannt: Tumoren, schwerwiegende Autoimmunphänomene und chronische Atemwegserkrankungen (Bronchiektasie).
Durch die Einführung der IVIG-Therapie hat sich die Prognose der Erkrankung in den letzten Jahrzehnten wesentlich verbessert.
Quellen
- ↑ Ulrich Salzer et al.: Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. In: Nature Genetics. Bd. 37, 2005, S. 820-828, ISSN 1061-4036
Literatur
- Mark Ballow: Primary immunodeficiency disorders: Antibody deficiency. In: J Allergy Clin Immunol. Bd. 109, Nr. 4, 2002, S. 581-591, ISSN 0091-6749
- Lennart Hammarström, Igor Vorechovský und David B. Webster: Selective IgA Deficiency (SIgAD) and common variable immunodeficiency (CVID). In: Clin Exp Immunol. Bd. 120, 2000, S. 225-231, ISSN 0009-9104
- Hans-Hartmut Peter, Werner J. Pichler (Hrsg.): Klinische Immunologie. 2. Auflage. Urban und Schwarzenberg, München; Wien; Baltimore 1996, ISBN 3-541-14892-6
- Fred S. Rosen et al.: Primary Immunodeficiency Diseases. Report of an IUIS Scientific Committee. In: Clin Exp Immunol. Bd. 118 (Suppl. 1), 1999, S. 1-28, ISSN 0009-9104
Weblinks
- Informationen des ImmunDefektCentrums der Charité
- Patientenmerkblatt der Arbeitsgemeinschaft Pädiatrische Immunologie
- Ausführlicher Leitfaden der Medizinischen Universitätsklinik Freiburg (PDF; 1,07 MB)
- Ausführlicher Artikel bei emedicine.com (engl.)
- Selbsthilfegruppe "DSAI - Deutsche Selbsthilfe Angeborene Immundefekte e.V."

Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.