- Ehlers-Danlos-Syndrom
-
Klassifikation nach ICD-10 Q79.6 Ehlers-Danlos-Syndrom ICD-10 online (WHO-Version 2011) Das Ehlers-Danlos-Syndrom (EDS) ist eine Gruppe von genetischen Bindegewebserkrankungen, die gekennzeichnet sind durch eine erhöhte Elastizität der Haut und ungewöhnliche Zerreißbarkeit derselbigen.
Inhaltsverzeichnis
Pathogenese
Aufgrund der hohen individuellen Variabilität der Erkrankung sowie unterschiedlicher genetischer Erbgänge werden seit einigen Jahren sechs Formen des Ehlers-Danlos-Syndroms unterschieden:
- Klassischer Typ (Typ I und II): stark überdehnbare und leicht verletzbare Haut, Hämatomneigung, abnorme Wundheilung, starke Überbeweglichkeit der Gelenke, innere Organe und Gefäße mit betroffen (Symptome bei Typ II wie bei Typ I, nur geringer ausgeprägt)
- Hypermobiler Typ (Typ III): geringe Beteiligung der Haut, ausgeprägte Überbeweglichkeit der Gelenke
- Vaskulärer Typ (Typ IV): dünne, durchscheinende Haut, ausgeprägte Hämatomneigung, Überbeweglichkeit der kleinen Gelenke, Beteiligung der inneren Organe und Gefäße
- Kyphoskoliotischer Typ (Typ VI): Überdehnbarkeit der Haut mittel bis stark, abnorme Wundheilung, starke Überbeweglichkeit der Gelenke, Augenbeteiligung, Beteiligung der inneren Organe[1]
- Arthrochalasie Typ (Typ VII A/B): Überdehnbarkeit der Haut (gering bis mittel), dünne Haut, Hüftluxation, ausgeprägte Überbeweglichkeit der Gelenke
- Dermatosparaxis Typ (Typ VII C): Haut sehr schlaff, deutliche Überbeweglichkeit der Gelenke, Beteiligung der inneren Organe
Allen gemeinsam ist eine Synthesestörung beziehungsweise mangelhafte oder fehlende Ausbildung der Kollagenvernetzung.
Bei Patienten mit vaskulärem Typ des Ehlers-Danlos-Syndroms wurden Mutationen im Gen des Rezeptors für Transforming Growth Factor beta nachgewiesen.[2]
Epidemiologie
Die autosomal-dominant vererbten Formen Typ I und II machen circa 80 % aller Erkrankungen aus.
Symptomatik
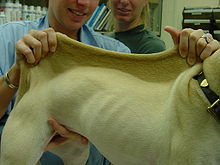 Ehlers-Danlos-Syndrom bei einem Hund
Ehlers-Danlos-Syndrom bei einem Hund
Typische Symptome beim Ehlers-Danlos-Syndrom sind die Hyperelastizität und die ungewöhnliche Zerreißbarkeit der Haut. Die Gelenke sind überstreckbar und neigen zu Luxationen. Nach leichten Verletzungen bleiben fischmaulartige Narben zurück. Weitere Symptome sind Kyphoskoliose, Mitralklappenprolaps, Aortenaneurysma und hämorrhagische Diathese. Einige EDS-Patienten leiden auch unter funktionellen Störungen der inneren Organe. Oft kommt es zu Eingeweidebrüchen (Hernien), meistens im Bereich des Dünndarmes.
Diagnostik
Das Ehlers-Danlos-Syndrom wird durch eine Anamnese und klinische Untersuchung diagnostiziert. Hauptbestandteil der Untersuchung ist eine Hautbiopsie mit Kollagenanalyse mittels Elektronenmikroskopie. Zur besseren Typisierung sind eigentlich weitergehende genetische Untersuchungen nötig, werden aber nicht realisiert, da die Kosten hierfür von den Krankenkassen nicht getragen werden und die Patienten die hohen Summen oft nicht aufbringen können.
Therapie
Eine randomisiert-kontrollierte Studie mit rund 50 Patienten über fünf Jahre kam zu dem Schluss, dass eine Betablockertherapie mit Celiprolol das Risiko von Gefäßkomplikationen auf die Hälfte senkt. Die Autoren der Studie sprachen sich ob der geringen Nebenwirkungen für eine generelle Therapieempfehlung aus.[3]
Prognose
Menschen mit Ehlers-Danlos-Syndrom haben in der Regel eine normale Lebenserwartung. Da es gehäuft zum Auftreten von Herz-Kreislauf-Erkrankungen kommt, ist eine regelmäßige kardiologische Abklärung notwendig. Eine regelmäßige augenärztliche Überwachung ist ebenfalls notwendig, da es zur Ablösung der Netzhaut und damit zur Erblindung kommen kann. Die Muskulatur sollte unbedingt gut trainiert werden, um die überdehnbaren Gelenke zu stabilisieren. Gut geeignet sind dazu Gymnastik, Radfahren oder leichter Kraftsport mit langsamen Bewegungen. Gelenkbelastende Bewegungsarten (Joggen) sind ungeeignet.
Geschichte
Nachdem sich im 18. Jahrhundert Fallberichte häuften, die meist in dermatologischen Zeitschriften veröffentlicht wurden, erfolgte die erste komplette wissenschaftliche Beschreibung 1891 durch einen russischen Mediziner. Benannt ist das Syndrom nach den beiden Dermatologen Edvard Ehlers (1863–1937) und Henri-Alexandre Danlos (1844–1912), die Anfang des 20. Jahrhunderts Studien auf dem Gebiet durchführten.
Zurzeit sind in Deutschland etwa 500 Menschen mit EDS bekannt, es wird jedoch mit einer hohen Dunkelziffer gerechnet.
Weblinks
Einzelnachweise
- ↑ Germain DP: Ehlers-Danlos syndrome type IV. Orphanet J Rare Dis. 2007 Jul 19;2:32. PMID 17640391
- ↑ Bart L. Loeys et. al: Aneurysm Syndromes Caused by Mutations in the TGF-ß Receptor. In: New England Journal of Medicine. Nr. 355, 2006, S. 788-798 (Abstract).
- ↑ Ong KT, Perdu J, De Backer J, Bozec E, Collignon P, et alia : Effect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: a prospective randomised, open, blinded-endpoints trial., Lancet. 2010 Sep 6 EPub ; PMID 20825986

Bitte den Hinweis zu Gesundheitsthemen beachten!
Wikimedia Foundation.