- Ewing-Sarkom
-
Klassifikation nach ICD-10 C40 Bösartige Neubildung des Knochens und des Gelenkknorpels der Extremitäten C41 Bösartige Neubildung des Knochens und des Gelenkknorpels sonstiger und nicht näher bezeichneter Lokalisationen ICD-10 online (WHO-Version 2011) 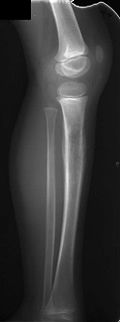 Röntgenaufnahme eines Ewing-Sarkoms der Tibia (Schienbein) bei einem Kind
Röntgenaufnahme eines Ewing-Sarkoms der Tibia (Schienbein) bei einem Kind
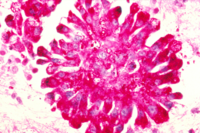 Metastatische Ewing-Sarkom-Zellen
Metastatische Ewing-Sarkom-ZellenBeim ossären Ewing-Sarkom handelt es sich um die zweithäufigste Art von Knochenkrebs. Es wurde zuerst vom US-amerikanischen Pathologen James Ewing beschrieben.
Die Erkrankung tritt bei Kindern und Jugendlichen (meistens Kaukasier) im Alter von fünf bis 27 Jahren auf (mit einer Häufung bei 10- bis 15-jährigen männlichen Jugendlichen) und ist extrem maligne. Häufig sind die Diaphysen der langen Röhrenknochen wie Oberschenkelknochen und Tibia betroffen. Es kommen jedoch auch Tumoren im Bereich des Schultergürtels und des Beckens vor. Es existiert zudem eine muskuläre Variante, die gemeinhin an den gleichen Stellen auftritt, aber statt des Knochens eben eine Muskelgruppe befällt. Hier treten ebenfalls starke Schmerzen und zudem z. B. massive Muskelkrämpfe im betroffenen Bereich auf.
Der Krebs wuchert sehr schnell und streut in andere Knochen und in die Lunge. Das Ewing-Sarkom gehört mit dem Primitiv neuroektodermalem Tumor zur Familie der Ewing-Tumoren, die durch ews/ets-Translokationen charakterisiert sind.[1][2]
Inhaltsverzeichnis
Symptome
Die Patienten weisen zu Beginn oft Beschwerden ähnlich einer Osteomyelitis auf. Im Vordergrund stehen Fieber, BSG-Erhöhung, erhöhte Werte von Leukozyten und CRP. Nicht selten zeigen die Patienten eine lokale Schwellung über dem betroffenen Knochen sowie Überwärmung und Schmerzen.
Diagnose
Typischerweise sind auf dem Röntgenbild mottenfraßartige Knochendestruktionen und ein Periostsporn (Codman-Dreieck) zu erkennen. Eine zwiebelschalenartige Periostverkalkung ist nur in etwa 20 % der Fälle zu finden. Bei Verdacht auf ein Ewing-Sarkom sichert letztlich eine Probebiopsie und deren pathologische Aufarbeitung die Diagnose.
Behandlung
Die Behandlung besteht aus einer Kombination aus Operation, intensiver Bestrahlungs- und Chemotherapie. Präoperativ wird eine Chemotherapie zur Tumorreduktion durchgeführt; zusätzlich verringert sich dadurch das Risiko, während der Operation eine Metastasierung zu begünstigen. Der Tumor muss operativ radikal reseziert werden, bestenfalls mit einem „Sicherheitsabstand“ von 5 cm im Gesunden. Der dadurch entstehende Knochendefekt wird je nach Situation durch ein Osteosyntheseverfahren versorgt. Nach der Operation erfolgt ein erneuter Zyklus Chemotherapie und Bestrahlung, jeweils den aktuellen Empfehlungen und Richtlinien entsprechend. Die Behandlung erfolgt üblicherweise gemäß dem Protokoll der Studie EURO-E.W.I.N.G. 99 (EUROpean Ewing tumour Working Initiative of National Groups – Ewing Tumour Studies 1999).
Prognose
Die Prognose ist maßgeblich abhängig von der Tumorausbreitung bei Diagnosestellung; die oft bereits vorhandenen Fernmetastasen verschlechtern die Prognose deutlich. Über alle Stadien hinweg haben die Patienten eine durchschnittliche 5-Jahres-Überlebensrate von etwa 50 %.
Entscheidende Faktoren für Überlebenswahrscheinlichkeit von Kindern mit Ewing-Sarkom sind Metastasierung, Tumorgröße und Tumorlokalisation. Die geschätzte Überlebenswahrscheinlichkeit von 15 Jahren nach Diagnosestellung in den Therapiestudien CESS 81, 86, 91P und EICESS 91 (1981–1997) beträgt für Patienten ohne Metastasen 61 % +/-5 %, die des rückfallfreien Überlebens 55 % +/-5 %. Sind bei Diagnosestellung bereits Metastasen vorhanden, verschlechtert sich die Prognose wesentlich. Hier liegt die 15-Jahres-Überlebenswahrscheinlichkeit bei 26 % +/-6 %.
Quellen
- ↑ Delattre O et al.:The Ewing family of tumors - a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med (1994) 331:294-299.
- ↑ Biology of EWS/ETS fusions in Ewing's family tumors. Oncogene (2001), Vol 20, 40, p. 5747–5754
Weblinks
- Nicht-aktualisierte Leitlinie zur Behandlung des Ewing-Sarkom im Internet Archive
- http://www.kinderkrebsinfo.de/Ewing-Sarkom_Kurzinfo
- Leitlinien (PDF)

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Krebserkrankung
- Kinderonkologie

Wikimedia Foundation.